The diagnosis of peripheral neuropathies can be frustrating, time consuming and costly. Careful clinical and electrodiagnostic assessment, with attention to the pattern of involvement and the types of nerve fibers most affected, narrows the differential diagnosis and helps to focus the laboratory evaluation. An algorithmic approach to the evaluation and differential diagnosis of a patient with peripheral neuropathy is presented, based on important elements of the clinical history and physical examination, the use of electromyography and nerve conduction studies, autonomic testing, cerebrospinal fluid analysis and nerve biopsy findings. The underlying cause of axonal neuropathies can frequently be treated; demyelinating neuropathies are generally managed with the assistance of a neurologist.
The incidence of peripheral neuropathy is not known, but it is a common feature of many systemic diseases. Diabetes and alcoholism are the most common etiologies of peripheral neuropathy in adults living in developed countries. The primary worldwide cause of treatable neuropathy is leprosy.1 Neuropathies associated with human immunodeficiency virus (HIV) infection account for an increasing number of cases. Peripheral neuropathy has numerous other causes, including hereditary, toxic, metabolic, infectious, inflammatory, ischemic and paraneoplastic disorders. The number of peripheral neuropathies for which an etiology cannot be found despite extensive evaluation ranges from 13 to 22 percent.2,3 Many undiagnosed patients (up to 42 percent) are found, after a careful family history and examination of kin, to have a familial neuropathy.2
The evaluation of a peripheral neuropathy can be time-consuming and costly. A systematic approach based on a careful clinical and electrodiagnostic assessment can help narrow the possibilities and tailor the laboratory evaluation to a specific differential diagnosis.
Anatomy
The peripheral nerves include the cranial nerves (with the exception of the second), the spinal nerve roots, the dorsal root ganglia, the peripheral nerve trunks and their terminal branches, and the peripheral autonomic nervous system. By convention, the motor neurons and their diseases are considered separately.
Nerves are composed of different types of axons. Large, myelinated axons include motor axons and the sensory axons responsible for vibration sense, proprioception and light touch. Small myelinated axons are composed of autonomic fibers and sensory axons and are responsible for light touch, pain and temperature. Small, unmyelinated axons are also sensory and subserve pain and temperature. Neuropathies involving primarily the latter two fiber types are called small-fiber neuropathies.
Clinically, large-fiber neuropathies can be distinguished from small-fiber neuropathies during neurologic testing: large fibers carry sensation for vibration and proprioception, while small fibers carry sensation for pain and temperature. Sensation for light touch is carried by both large and small nerve fibers.
Pathophysiology
Although peripheral neuropathy has multiple etiologies, the nerve has a limited number of ways to respond to injury.4,5 The damage can occur at the level of the axon (i.e., axonopathy). A disruption of the axons (e.g., trauma) results in degeneration of the axon and the myelin sheath distal to the site of the injury (i.e., Wallerian degeneration). In most toxic and metabolic injuries, the most distal portion of the axons degenerates, with concomitant breakdown of the myelin sheath (known as “dying-back,” or length-dependent, neuropathy).
Neuronopathies occur at the level of the motor neuron or dorsal root ganglion, with subsequent degeneration of their peripheral and central processes. Because the injury is at the level of the cell body, recovery is often incomplete.
Myelinopathies occur at the level of the myelin sheath and can be inflammatory or hereditary. In acquired demyelinating neuropathies, the injury is often patchy or segmental. Because the axons are relatively spared, recovery is often rapid (weeks to months) and complete. Hereditary abnormalities of myelin are usually diffuse, with a slowly progressive course.
Diagnostic Approach
The differential diagnosis of peripheral neuropathy is significantly narrowed by a focused clinical assessment that addresses several key issues (Figure 1). The first issue is, does the patient actually have a neuropathy? Causes of generalized weakness include motor neuron disease, disorders of the neuromuscular junction and myopathy. Peripheral neuropathy can also be mimicked by myelopathy, syringomyelia or dorsal column disorders, such as tabes dorsalis. Hysterical symptoms can sometimes mimic a neuropathy.
FIGURE 1.
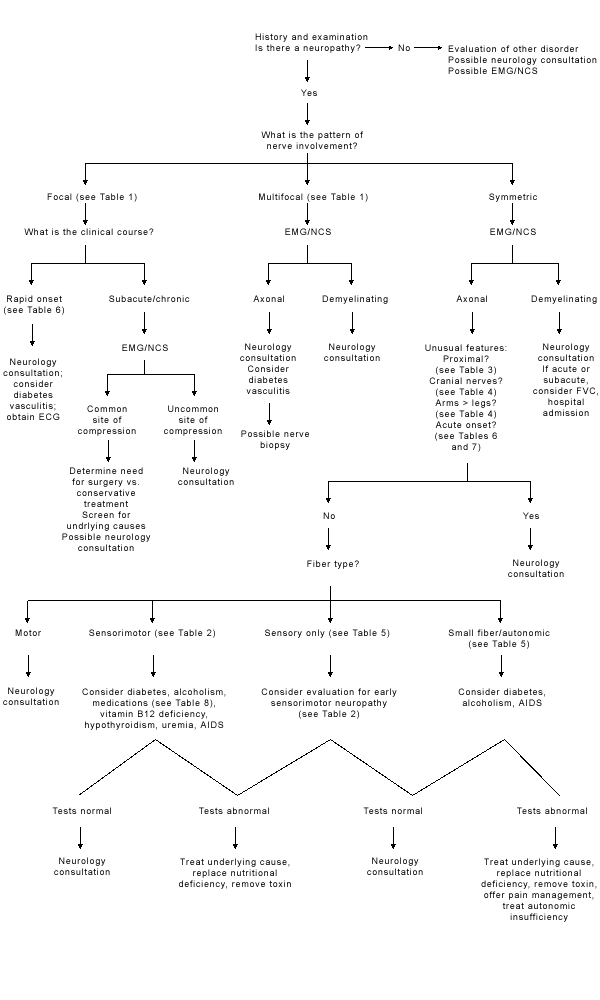
Algorithm for evaluation of a patient with a peripheral neuropathy. (ECG = electrocardiogram; EMG/NCS = electron microscopy/nerve conduction studies; AIDS = acquired immunodeficiency syndrome; FVC = forced vital capacity)
It is useful to determine the pattern of involvement. Is the neuropathy focal, multifocal or symmetric? Focal neuropathies include common compressive neuropathies such as carpal tunnel syndrome, ulnar neuropathy at the elbow or peroneal neuropathy at the fibular head6,7 (Table 1).8 A multifocal neuropathy suggests a mononeuritis multiplex that may be caused, for example, by vasculitis or diabetes (Table 1).8
TABLE 1 Neuropathies by Pattern of Involvement
| Focal | |
| Entrapment | |
| Common sites of compression | |
| Myxedema | |
| Rheumatoid arthritis | |
| Amyloidosis | |
| Acromegaly | |
| Compressive neuropathies | |
| Trauma | |
| Ischemic lesions | |
| Diabetes mellitus | |
| Vasculitis | |
| Leprosy | |
| Sarcoidosis | |
| Neoplastic infiltration or compression | |
| Multifocal | |
| Diabetes mellitus | |
| Vasculitis | |
| Polyarteritis nodosa | |
| Systemic lupus erythematosus | |
| Sjögren's syndrome | |
| Sarcoidosis | |
| Leprosy | |
| HIV/AIDS | |
| Multifocal variant of CIDP | |
| Hereditary predisposition to pressure palsies | |
HIV = human immunodeficiency virus; AIDS = acquired immunodeficiency syndrome; CIDP = chronic inflammatory demyelinating polyradiculoneuropathy.
Information from Thomas PK, Ochoa J. Symptomatology and differential diagnosis of peripheral neuropathy. In: Dyck PJ, Thomas PK, eds. Peripheral neuropathy. Philadelphia: Saunders, 1993:749–74.
If the neuropathy is symmetric, is it proximal or distal? Most toxic and metabolic neuropathies present as a distal symmetric or dying-back process (Table 2).9 Proximal sensory neuropathies are rare and include porphyria.6 Predominantly motor neuropathies are often proximal and include acquired inflammatory neuropathies such as Guillain-Barré syndrome8,9 (Table 3).8 An exception is lead neuropathy, which initially affects motor fibers in radial and peroneal distributions.
TABLE 3 Proximal Symmetric Motor Polyneuropathies
| Guillain-Barré syndrome |
| Chronic inflammatory demyelinating polyradiculoneuropathy |
| Diabetes mellitus |
| Porphyria |
| Osteosclerotic myeloma |
| Waldenstrom's macroglobulinemia |
| Monoclonal gammopathy of undetermined significance |
| Acute arsenic polyneuropathy |
| Lymphoma |
| Diphtheria |
| HIV/AIDS |
| Lyme disease |
| Hypothyroidism |
| Vincristine (Oncovin, Vincosar PFS) toxicity |
HIV = human immunodeficiency virus; AIDS = acquired immunodeficiency syndrome.
Information from Thomas PK, Ochoa J. Symptomatology and differential diagnosis of peripheral neuropathy. In: Dyck PJ, Thomas PK, eds. Peripheral neuropathy. Philadelphia: Saunders, 1993:749–74.
A limited number of neuropathies involve the cranial nerves (Table 4).8 Guillain-Barré syndrome frequently involves the facial nerves. Another uncommon pattern is greater involvement of the arms than the legs (Table 4).8 Leprosy tends to involve cutaneous nerves in cooler areas of the body, such as the tip of the nose, the pinna of the ear and the volar surfaces of the arms.
TABLE 4 Neuropathies with Less Common Patterns of Involvement
| Neuropathies with cranial nerve involvement |
| Diabetes mellitus |
| Guillain-Barré syndrome |
| HIV/AIDS |
| Lyme disease |
| Sarcoidosis |
| Neoplastic invasion of skull base or meninges |
| Diphtheria |
| Neuropathies predominant in upper limbs |
| Guillain-Barré syndrome |
| Diabetes mellitus |
| Porphyria |
| Hereditary motor sensory neuropathy |
| Vitamin B12 deficiency |
| Hereditary amyloid neuropathy type II* |
| Lead neuropathy |
HIV = human immunodeficiency virus; AIDS = acquired immunodeficiency syndrome.
*—Carpal tunnel syndrome resulting from amyloid deposits in the flexor retinaculum.
Information from Thomas PK, Ochoa J. Symptomatology and differential diagnosis of peripheral neuropathy. In: Dyck PJ, Thomas PK, eds. Peripheral neuropathy. Philadelphia: Saunders, 1993:749–74.
Neuropathies can be categorized according to the fiber type that is primarily involved. Most toxic and metabolic neuropathies are initially sensory and later may involve the motor fibers (Table 2).9 Pure sensory neuropathies or neuronopathies can result from drug toxicity (e.g., thalidomide, cisplatin [Platinol]), paraneoplastic syndromes or nutritional deficiencies (Table 5).8,9 Primarily motor neuropathies include Guillain-Barré syndrome8,9 (Table 38 ). Alcoholism and diabetes can both cause small-fiber, painful neuropathies (Table 5).8,9 Autonomic involvement occurs in many small-fiber neuropathies but can also occur in Guillain-Barré syndrome and is sometimes life-threatening (Table 5).8,9 It is important to distinguish whether the neuropathy is axonal, demyelinating, or both. This differentiation is best achieved using nerve conduction studies (NCS) and electromyography (EMG).
TABLE 5 Comparative Patterns of Neuropathies and Neuronopathies by Fiber Type
| Pure sensory neuropathies and neuronopathies |
| Paraneoplastic |
| Medications (see Table 8) |
| Carcinomatous sensory neuronopathy |
| Lymphomatous sensory neuronopathy |
| Sjögren's syndrome |
| Paraproteinemias |
| Nonsystemic vasculitic neuropathy |
| Idiopathic sensory neuronopathy |
| Styrene-induced peripheral neuropathy |
| Primary biliary cirrhosis |
| Crohn's disease |
| Chronic gluten enteropathy |
| Vitamin E deficiency |
| Hereditary sensory neuropathy types I and IV |
| Friedreich's ataxia |
| Small-fiber neuropathies |
| Leprosy |
| Diabetes mellitus |
| Alcoholic neuropathy |
| Amyloidosis |
| AIDS |
| Hereditary |
| Neuropathies with autonomic involvement |
| Diabetic neuropathy |
| Amyloidosis |
| Porphyria |
| Paraneoplastic neuropathy |
| Lymphoma |
| Thallium, arsenic, mercury toxicity |
| Thiamine deficiency |
| Vincristine (Oncovin, Vincosar PFS) toxicity |
| Guillain-Barré syndrome |
| Alcoholic neuropathy |
| Acute pandysautonomia |
| HIV/AIDS |
AIDS = acquired immunodeficiency syndrome; HIV = human immunodeficiency virus.
Information from Donofrio PD, Albers JW. AAEM minimonograph #34. Polyneuropathy: classification by nerve conduction studies and electromyography. Muscle Nerve 1990;13:889–903, and Thomas PK, Ochoa J. Symptomatology and differential diagnosis of peripheral neuropathy. In: Dyck PJ, Thomas PK, eds. Peripheral neuropathy. Philadelphia: Saunders, 1993:749–74.
Diabetes, HIV infection and alcoholism can cause several patterns of neuropathy. They most commonly cause a distal, symmetric axonal sensorimotor neuropathy. The second most common presentation in these conditions is a small-fiber, painful neuropathy. Involvement of autonomic fibers is common in diabetes but less common in acquired immunodeficiency syndrome (AIDS) or alcoholism. These three patterns of neuropathy occur only in the AIDS stage of HIV infection. Medications used to treat HIV infection, such as didanosine (ddI; Videx) and zalcitamine (ddC; Hivid) also cause a distal symmetric axonal sensorimotor neuropathy.
Diabetes infrequently causes multifocal neuropathies including the cranial nerves, an asymmetric proximal motor neuropathy (diabetic amyotrophy) or a symmetric proximal motor neuropathy. HIV seroconversion rarely can be associated with an acute or chronic demyelinating neuropathy. In AIDS, polyradiculopathy or mononeuritis multiplex associated with cytomegalovirus infection can also occur.
History
The temporal course of a neuropathy varies, based on the etiology (Tables 6 and 7).8,9 With trauma or ischemic infarction, the onset will be acute, with the most severe symptoms at onset. Inflammatory and some metabolic neuropathies have a subacute course extending over days to weeks. A chronic course over weeks to months is the hallmark of most toxic and metabolic neuropathies. A chronic, slowly progressive neuropathy over many years occurs with most hereditary neuropathies or with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Neuropathies with a relapsing and remitting course include Guillain-Barré syndrome.
TABLE 6 Neuropathies with Abrupt/Rapid Onset
| Ischemic neuropathies | |
| Polyarteritis nodosa | |
| Rheumatoid arthritis | |
| Diabetes mellitus | |
| Cranial neuropathies | |
| Diabetic amyotrophy | |
| Nerve compression | |
| Hemorrhage | |
| Swelling within a restricted anatomic compartment (e.g., anterior tibial syndrome) | |
| Direct external compression | |
| Penetrating wounds | |
| Thermal injury | |
| Iatrogenic (e.g., injection into nerves) | |
Information from Donofrio PD, Albers JW. AAEM minimonograph #34. Polyneuropathy: classification by nerve conduction studies and electromyography. Muscle Nerve 1990;13:889–903, and Thomas PK, Ochoa J. Symptomatology and differential diagnosis of peripheral neuropathy. In: Dyck PJ, Thomas PK, eds. Peripheral neuropathy. Philadelphia: Saunders, 1993:749–74.
TABLE 7 Differential Diagnosis of Neuropathies by Clinical Course
| Acute onset (within days) | Subacute onset (weeks to months) | Chronic course/insidious onset | Relapsing/remitting course |
|---|---|---|---|
| Guillain-Barré syndrome | Maintained exposure to toxic agents/medications | Hereditary motor sensory neuropathies | Guillain-Barré syndrome |
| Acute intermittent porphyria | Persisting nutritional deficiency | Dominantly inherited sensory neuropathy | CIDP |
| Critical illness polyneuropathy | Abnormal metabolic state | CIDP | HIV/AIDS |
| Diphtheric neuropathy | Paraneoplastic syndrome | Toxic | |
| Thallium toxicity | CIDP | Porphyria |
CIDP = chronic inflammatory demyelinating polyradiculoneuropathy; HIV = human immunodeficiency virus; AIDS = acquired immunodeficiency syndrome.
Information from Thomas PK, Ochoa J. Symptomatology and differential diagnosis of peripheral neuropathy. In: Dyck PJ, Thomas PK, eds. Peripheral neuropathy. Philadelphia: Saunders, 1993:749–74.
The symptoms and signs of neuropathy not only suggest the presence of neuropathy but may also indicate the type of axons involved. Ischemic neuropathies often have pain as a prominent feature. Small-fiber neuropathies often present with burning pain, lightning-like or lancinating pain, aching, or uncomfortable paresthesias (dysesthesias). Patients may complain of pain with innocuous stimuli such as sheets rubbing over their feet (allodynia). They may also describe a tight, band-like sensation around the ankles or wrists. Sensory symptoms include tingling or paresthesias, increased sensation in affected areas (hypesthesia), and numbness or reduced sensation. Dying-back (distal symmetric axonal) neuropathies initially involve the tips of the toes and progress proximally in a stocking-glove distribution. Multifocal neuropathies, such as mononeuritis multiplex caused by polyarteritis nodosa, may result in sensory abnormalities in specific nerve or root distributions.
Motor symptoms such as weakness and wasting also commence distally in a dying-back neuropathy. Common complaints are tripping on the toes and loss of grip strength. The patient may have cramps or fasciculations. Peripheral neuropathy can present as restless leg syndrome. Proximal involvement may result in difficulty climbing stairs, getting out of a chair, lifting and swallowing, and in dysarthria.
The clinical assessment should include a careful past medical history, looking for systemic diseases that can be associated with neuropathy, such as diabetes or hypothyroidism. Many medications can cause a peripheral neuropathy (Table 8),10 typically a distal symmetric axonal sensorimotor neuropathy. Detailed enquiries about drug and alcohol use, as well as exposure to heavy metals and solvents, should be pursued. All patients should be questioned regarding HIV risk factors, foreign travel (leprosy), diet (nutrition), vitamin use (especially B6) and the possibility of a tick bite (Lyme disease). A detailed family history should include inquiries as to the presence of hammer toes, high arches, weak ankles, gait abnormalities or “muscular dystrophy,” that would suggest a longstanding or hereditary neuropathy. The review of systems may provide clues regarding other organ involvement and the presence of an underlying malignancy.
Physical Examination
A cranial nerve examination can provide evidence of mononeuropathies or proximal involvement. In addition, a funduscopic examination may show abnormalities such as optic pallor, which can be present in leukodystrophies and vitamin B12 deficiency. Direct strength testing of muscles enervated by cranial nerves V, VII, IX/X, XI and XII is important, as mild bilateral weakness can be missed by observation only. The motor examination includes a search for fasciculations or cramps, or loss of muscle bulk. Tone is normal or reduced. The pattern of weakness helps narrow the diagnosis: symmetric or asymmetric, distal or proximal, and confined to a particular nerve, plexus or root level, as indicated in Tables 1 through 3.
In a patient with a distal symmetric sensorimotor neuropathy, the sensory examination shows reduced sensitivity to light touch, pin-prick and temperature in a stocking-and-glove distribution. Vibration and position sense are reduced in the distal legs prior to involvement of the arms. In patients with severe loss of position sense, there may be athetoid movement of the fingers or arms when the eyes are closed (pseudoathetosis) or a Romberg sign. Patients with mononeuritis multiplex may have sensory loss in specific nerve distributions.
Deep tendon reflexes are reduced or absent. A bilateral foot drop may result in a steppage gait in which the patient must lift the knees very high in order to clear the toes. Proximal weakness results in an inability to squat or to rise unassisted from a chair.
Severe, longstanding neuropathy can result in trophic changes including pes cavus, kyphoscoliosis, loss of hair in affected areas or ulceration. Radiographic examination of limbs may show loss of bone density, thinning of phalanges, pathologic fractures or neuropathic arthropathy. Trophic changes are most prominent in diabetes, amyloid neuropathy, leprosy, hereditary motor sensory neuropathy (HMSN) with prominent sensory involvement, and hereditary sensory neuropathy. Nerve thickening can be palpated in leprosy, HMSN type 1 and amyloid neuropathy.
The general physical examination can provide evidence of orthostatic hypotension without a compensatory rise in heart rate when autonomic fibers are involved. Respiratory rate and vital capacity should be evaluated in Guillain-Barré syndrome to assess for respiratory compromise. The presence of lymphadenopathy, hepatomegaly or splenomegaly, and skin lesions may provide evidence of systemic disease. Pale transverse bands in the nail beds, parallel to the lunula (Mees' lines), suggest arsenic poisoning.
Laboratory Evaluation
EMG and nerve conduction studies (NCS) are often the most useful initial laboratory studies in the evaluation of a patient with peripheral neuropathy.4,5,9,11,12 They can confirm the presence of a neuropathy and provide information as to the type of fibers involved (motor, sensory, or both), the pathophysiology (axonal loss versus demyelination) and a symmetric versus asymmetric or multifocal pattern of involvement. Sensory axonal neuropathy and sensory neuronopathy have similar electrodiagnostic features and are considered together. The differential diagnosis of different types of peripheral neuropathy can be divided using electrophysiologic criteria.7
Axon loss results in loss of amplitude of nerve action potentials, and evidence of denervation is found on needle examination of affected muscles. Myelin loss results in slowed conduction velocities, prolonged distal latencies, conduction block, temporal dispersion and prolonged minimum F-wave latencies. The limitations of EMG/NCS should be taken into account when interpreting the findings. We have no reliable means of studying proximal sensory nerves. NCS results can be normal in patients with small-fiber neuropathies, and lower extremity sensory responses can be absent in normal elderly patients. EMG/NCS are not substitutes for a good clinical examination.
Subsequent studies should be tailored to the most likely diagnostic possibilities, and to the acuteness and severity of the neuropathy. With an acute progressive neuropathy, a neurologic consultation early in the course of the evaluation is essential. Further evaluation of these patients includes EMG/NCS, lumbar puncture, chest radiograph, electrocardiogram and determination of forced vital capacity. More indolent neuropathies can be evaluated in a cost-efficient manner by the family physician.
The most common presentation is that of a distal symmetric sensorimotor neuropathy. Initial evaluation should include fasting serum glucose, glycosylated hemoglobin, blood urea nitrogen, creatinine, complete blood cell count, erythrocyte sedimentation rate, urinalysis, vitamin B12 and thyrotropin stimulating hormone levels. Neurologic assessment may be warranted if the initial evaluation does not produce a diagnosis.
Autonomic studies include determination of heart rate variation with respiration, heart rate response to standing/tilting, blood pressure response to sustained hand grip and a measure of sympathetic skin response. The results of these tests can provide objective evidence of autonomic insufficiency and a measure of small-fiber function.
The cerebrospinal fluid is useful in evaluation of myelinopathies and polyradiculopathies. An elevated total protein level with less than 5 white blood cells (albuminocytologic dissociation) is present in acquired inflammatory neuropathy (e.g., Guillain-Barré syndrome, CIDP). Other studies useful in specific clinical contexts are cytology (lymphoma) and special studies such as Lyme polymerase chain reaction and cytomegalovirus branched chain DNA (polyradiculopathy or mononeuritis multiplex in AIDS).
Nerve biopsy is only helpful in very specific cases to diagnose vasculitis, leprosy, amyloid neuropathy, leukodystrophies, sarcoidosis and, occasionally, CIDP. The sural nerve is the one most commonly selected for biopsy. Complications include infection, poor wound healing and painful dysesthesias. The biopsy should be performed and evaluated by an experienced surgeon and neuropathologist.
It can be difficult to document a small-fiber neuropathy because the only abnormalities on neurologic examination may be loss of pin-prick and temperature sensation in a distal distribution. EMG/NCS may be normal. Autonomic studies are only helpful if the autonomic fibers are involved. As a result, small-fiber neuropathy remains a primarily clinical diagnosis. The evaluation should include the most likely causes (i.e., diabetes, alcoholism, AIDS). If these studies are normal, a neurologic consultation is recommended.
