Of the uncommon anemias, “common” types include the anemia of renal disease, thalassemia, myelodysplastic syndrome and the anemia of chronic disease. These conditions may be suggested by the clinical presentation, laboratory test values and peripheral blood smear, or by failure of the anemia to respond to iron supplements or nutrient replacement. The principal cause of the anemia of renal disease is a decreased production of red blood cells related to a relative deficiency of erythropoietin. When treatment is required, erythropoietin is administered, often with iron supplementation. In the anemia of chronic disease, impaired iron transport decreases red blood cell production. Treatment is predominantly directed at the underlying condition. Since iron stores are usually normal, iron administration is not beneficial. Thalassemia minor results from a congenital abnormality of hemoglobin synthesis. The disorder may masquerade as mild iron deficiency anemia, but iron therapy and transfusions are often not indicated. In the myelodysplastic syndrome, blood cell components fail to mature, and the condition may progress to acute nonlymphocytic leukemia. The rate of progression depends on the subtype of myelodysplasia, but the leukemia is usually resistant to therapy.
Most cases of anemia, especially those related to vitamin B12, folate and iron deficiency, are diagnosed and appropriately treated by family physicians. When the underlying cause of anemia is obscure, a hematologic evaluation is often sought.
In 50 consecutive anemic patients who were referred to our institutions, the most common diagnoses of non-nutritional anemia were anemia of renal disease, anemia of chronic disease (ACD), thalassemia and myelodysplastic syndrome (Table 1). The salient features of these disorders, including pathophysiology, diagnosis and treatment, are reviewed in this article. Some important distinctive aspects are summarized in Table 2.
TABLE 1 Causes of Anemia in 50 Patients Referred to a Regional Center
| Diagnosis | Number of patients (%) | |
|---|---|---|
| Unrecognized iron deficiency | 11 (22) | |
| Anemia of chronic disease | 11 (22) | |
| Arthritis (4) | ||
| Malignancy (4) | ||
| Infection (3) | ||
| Anemia of renal disease | 6 (12) | |
| Thalassemia | 4 (8) | |
| Myelodysplasia | 5 (10) | |
| Miscellaneous | 10 (20) | |
| Liver disease (1) | ||
| Nutritional (2) | ||
| Hemolysis (2) | ||
| Unknown (5) | ||
| Spurious* | 3 (6) | |
*—No evidence of anemia was found.
Information from authors' unpublished study.
TABLE 2 Characteristics of Anemias
| Iron stores | ||||
|---|---|---|---|---|
| Cause or type | Ferritin | TIBC | Marrow Fe | Treatment |
| Iron deficiency | Low | High | Absent | Iron supplements; treat source of blood loss |
| Renal disease | Normal | Normal or low | Normal | Erythropoietin* |
| Anemia of chronic disease | Normal or high | Normal or low | Normal or increased | Underlying disease |
| Thalassemia minor | Normal | Normal | Normal | None |
| Myelodysplasia | Normal | Normal or low | Normal† | Supportive |
TIBC = total iron-binding capacity; Fe = iron.
*—May require adjunctive iron supplementation.
†—Exception: patients with refractory anemia with sideroblasts whose levels are markedly increased.
Unsuspected iron deficiency accounted for 22 percent of the referrals. Iron deficiency was under-recognized because of an insufficient history, a lack of stool examination for blood or poor responses to iron therapy because of continued blood loss or the suboptimal use of iron supplements. Although iron deficiency is important, this review focuses on the “common” uncommon anemias: that is, the anemias of renal disease and chronic disease, thalassemia and myelodysplasia.
Figure 1 illustrates the initial evaluation of anemia. This protocol relies on repeated laboratory testing to affirm the presence of anemia. In addition, the pathophysiologic mechanisms of anemia (red blood cell production defects and increased red blood cell destructive processes) are screened by reticulocyte counts, mean corpuscular volume (MCV) determinations and occult stool blood tests.
FIGURE 1. Initial Evaluation of Anemia
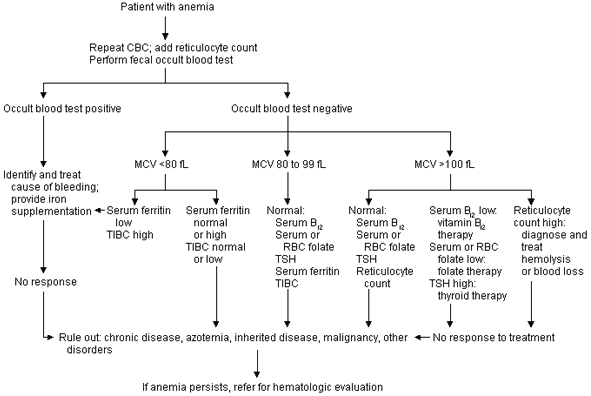
Protocol for the initial investigation of a patient with anemia. (CBC = complete blood count; MCV = mean corpuscular volume; TIBC = total iron-binding capacity; RBC = red blood cell; TSH = thyroid-stimulating hormone)
Anemia of Renal Disease
Anemia of renal disease occurs in both acute and chronic renal failure. Renal excretory dysfunction and endocrine abnormalities play pathophysiologic roles. This is demonstrated by the build-up of uremic toxins and decreased erythropoietin production, both of which adversely affect erythropoiesis. The accumulation of toxic metabolites, which are normally excreted by the kidneys, shortens the life span of circulating red blood cells. In fact, there is an inverse relationship between blood urea nitrogen (BUN) levels and red blood cell life span. More important, however, is the diminished renal production or the dysfunction of erythropoietin that results in decreased red blood cell production. Although the erythropoietin levels may be low, normal or high, they are inappropriately low when adjusted for the degree of anemia.1,2
The anemia of renal disease is usually normocytic and normochromic, but microcytosis can also occur. Hemoglobin concentrations may be mildly decreased with lesser degrees of azotemia or markedly diminished in chronic renal failure (hemoglobin levels as low as 5 to 7 g per dL [50 to 70 g per L]). A variety of morphologic abnormalities can be seen, such as burr cells in cases of uremia; microcytic, hypochromic red blood cells despite normal iron stores; or schistocytes and thrombocytopenia in cases of disseminated intravascular coagulation or thrombotic thrombocytopenic purpura.
Often serum iron levels and percentage of iron saturation are low, and sometimes iron replacement is erroneously given for the treatment of anemia of renal disease. However, these low values may reflect the role of iron as a negative acute-phase reactant rather than signifying iron deficiency. Although most acute-phase reactants are associated with increased levels of the reactant in question (such as the serum ferritin level), the serum iron and transferrin (total iron-binding capacity [TIBC]) levels decrease nonspecifically in response to various disease states.
Androgens were used in the past in an attempt to stimulate red blood cell production, but the administration of erythropoietin has effectively replaced this treatment. In patients with a relative deficiency of erythropoietin, both intravenous and subcutaneous administration of erythropoietin increases the reticulocyte count and red blood cell mass in a dose-related manner.3 Even when iron stores are adequate, iron supplementation is usually needed with erythropoietin administration because enhanced red blood cell production rapidly depletes the stores. Erythropoietin can be given three times per week, but higher dosages on a once-per-week basis may also be effective. Because erythropoietin therapy is expensive (Medicare reimburses at a rate of $1,500 per month), the decision to use it should be made on clinical grounds rather than on the basis of an absolute hemoglobin level.
Anemia of Chronic Disease
ACD occurs commonly, but it is frequently misunderstood, underdiagnosed or improperly treated. It is usually associated with infection, inflammation, malignancy or trauma. The predominant clinical findings reflect the underlying disorder.
The precise pathophysiologic mechanism of ACD is unclear. The anemia is related more to decreased red blood cell production than to increased destruction. There is an impairment in the transport of iron from iron storage sites, such as the liver and bone marrow, to red blood cells (Figure 2). Infection or inflammation may be responsible for the production of mediators (such as interleukin-1) that result in increased sequestration of iron in macrophages and hepatocytes. Additional factors are decreased levels of transferrin (TIBC) and competition with another transport protein (lactoferrin) that preferentially returns iron back to macrophages (Figure 2). Therefore, iron is present but inaccessible for use in the production of hemoglobin within erythrocytes. A poor response to or a relative decrease in erythropoietin production complicates the anemia.4 Finally, an unexplained slightly shortened red blood cell survival also contributes to the anemia.
FIGURE 2.
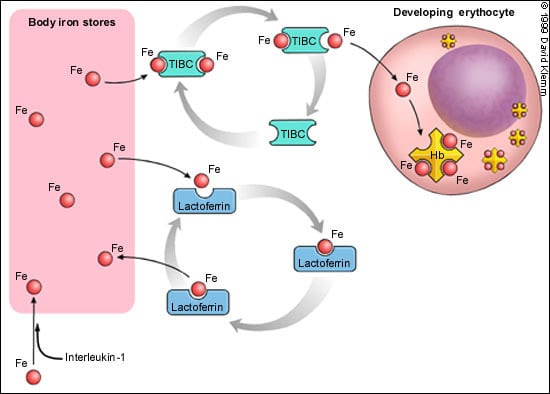
Diagram of iron kinetics from iron stores to developing red blood cell (RBC). Iron stores include the bone marrow, reticuloendothelial system (liver and spleen) and RBCs. Transferrin (total iron-binding capacity [TIBC]) transports iron (Fe) to developing erythrocytes. Iron is deposited in the RBC, and transferrin returns to storage sites to bind more Fe for transport. Lactoferrin is a competitor of transferrin; it takes Fe that is free and returns it to storage sites. Lactoferrin levels are elevated in anemia of chronic disease. Increases in interleukin-1 increase the sequestration of Fe in storage sites. (Hb = hemoglobin)
Although ACD may become marked, it is usually nonprogressive, with hemoglobin levels remaining above 9 g per dL (90 g per L). Erythrocytes are normocytic (MCU: 80 to 100 fL) or mildly microcytic (MCU: less than 80 fL). Often the peripheral blood smear is nearly normal, as shown in Figure 3.
FIGURE 3.
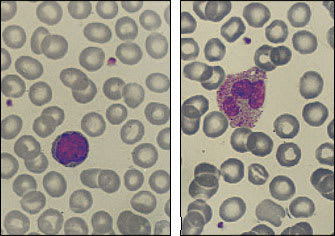
Sections of peripheral blood smears from two patients with anemia of chronic disease. (Left) Normal lymphocyte; normal red blood cell sizes approximate the diameter of a mature lymphocyte. The erythrocytes shown here are normal to mildly microcytic. (Right) Red blood cells here are also slightly microcytic; however, the polymorphonuclear leukocyte contains excessive granules (toxic granulation), which suggests an underlying infection or inflammation. (Oil immersion; × 1,000)
One of the characteristic laboratory features of ACD is a low serum iron level, often misinterpreted as iron deficiency. Patients with ACD usually have normal to increased body iron stores; therefore, ferritin levels may be normal or increased (Table 2). The interpretation of iron values may be complicated in patients with liver inflammation; iron and ferritin may be released from liver cells during inflammation and cause elevated values of serum iron and serum ferritin.5 In patients with ACD, bone marrow examinations are not necessary other than to confirm the adequacy of iron stores.
The treatment of ACD is directed at the underlying cause. Iron therapy is of no benefit. If necessary, transfusions or erythropoietin therapy can be considered when the degree of anemia poses cardiovascular problems. When ACD is associated with cancer, other factors may contribute to the anemia, such as transient bone marrow suppression from chemotherapy or bone marrow replacement by tumor. In patients with cancer, treatment with transfusions or erythropoietin is acceptable because of greater degrees of anemia and other complicating features such as bleeding, thrombocytopenia and relative deficiency of erythropoietin.6,7
Thalassemia
The frequency of thalassemia is dependent on the ethnic origins of the patient population. β-Thalassemia is well recognized in persons of Greek and Italian descent, whereas the α-thalassemic syndromes have an increased frequency in black, American Indian and Asian groups. Collectively, the thalassemias are among the most common inherited disorders.8
The pathophysiologic mechanism in thalassemia is related to an unbalanced synthesis of alpha chains (α-thalassemia) or beta chains (β-thalassemia). Normally, the synthesis of alpha and beta chains is balanced, resulting in normal hemoglobin A (α2β2). An unbalanced synthesis of either chain can lead to a failure in the matching of these chains and a defect in hemoglobinization within erythrocytes. As a result, red blood cell death can occur in the marrow (ineffective erythropoiesis), or the life span of the red blood cells that exit the marrow may be shortened.
Ordinarily, severe forms of thalassemia, such as β-thalassemia major, present in childhood with marked anemia, growth disturbances and jaundice, and are promptly diagnosed. Thalassemia minor, however, may go unrecognized into adulthood until a routine laboratory evaluation or an intercurrent problem prompts medical attention. These patients have mild to minimal anemia with hypochromic and microcytic red blood cells. Splenic enlargement may also be present. A family history of anemia may be the most important factor in the hematologic evaluation. β-Thalassemia minor is easily confirmed by hemoglobin electrophoresis, which shows an elevated hemoglobin A2 level, increased levels of hemoglobin F, or both.
The expression of α-thalassemia is more complex than that of β-thalassemia because two genes regulate alpha chain synthesis. No recognizable hemoglobin electrophoretic abnormalities occur with two forms of α-thalassemia (silent carrier and α-thalassemia trait). Hemoglobin H, a more aggressive form of α-thalassemia, shows excessive amounts of unbalanced beta chains, referred to as hemoglobin H.
A valuable clue to the diagnosis of either β- or α-thalassemia minor can be the presence of moderate peripheral blood smear abnormalities despite an insignificant anemia (Figure 4). In some patients, the diagnosis can be confirmed by a family study showing the presence of minimal anemia, microcytosis or splenomegaly.
FIGURE 4.
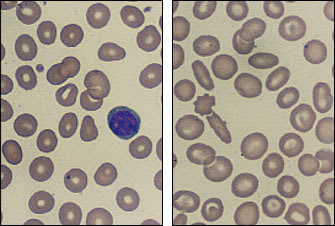
Peripheral blood smears from two patients with β-thalassemia minor. (Left) Note the microcytic cells with an occasional tear-shaped red blood cell. (Right) Target cells, oval shapes and microcytes. (Oil immersion; × 1,000)
The importance of diagnosing thalassemia minor lies in the genetic information obtained and in the recognition that these disorders (both α- and β-thalassemia) require little or no therapy. It is unusual for affected patients to need transfusions or marrow stimulants such as erythropoietin. At times, laboratory data may suggest iron deficiency, but iron supplementation is usually not needed unless the patient has excessive blood loss and confirmed iron deficiency. Unnecessary iron therapy is harmful because of iron overload and potential organ damage.
Myelodysplasia
During the past few decades, myelodysplasia has been variously labeled as hyperplastic bone marrow with bone marrow failure, refractory anemia, smoldering leukemia and preleukemia. The currently accepted term is “myelodysplastic syndrome,” an appropriate name because any of the marrow cell lines can be involved, maturation of the cell lines is dyspoietic and acute leukemia is not inevitable. This disease can be summarized as one in which abundant marrow lacks normal maturation of cellular components so that the delivery of mature cells into the peripheral blood is deficient.
It is difficult to determine the incidence of the myelodysplastic syndrome because the disease is not included as a diagnostic category in most tumor registries. The frequency of myelodysplasia appears to increase with advancing age. This disease should be considered in patients with megaloblastic anemia but without folic acid, vitamin B12 or thyroid abnormalities, as well as in patients who have normocytic or microcytic anemia without evidence of iron deficiency, thalassemia, renal disease or chronic disease. The presence of anemia combined with abnormalities of one or two of the other marrow cell lines is also suggestive of myelodysplastic syndrome. Abnormalities of red blood cell shape and size, polymorphonuclear leukocytes with hypersegmentation or Pelger-Huët nuclear anomaly should raise further suspicion of the disease. Some of these findings are seen in Figure 5. This hematologic disease can be monoclonal, and chromosomal studies may detect recognized deletions such as 5q(-) and 7q(-) or trisomy 8.9
FIGURE 5.
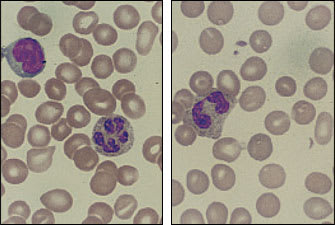
Peripheral smear from a patient with myelodysplasia. (Left) Hypersegmented polymorphonuclear leukocyte. Such cells can be seen in patients with myelodysplasia; however, they are nonspecific and can also appear in patients with folic acid and vitamin B12 deficiency, thyroid deficiency, myeloproliferative diseases, uremia and hereditary hypersegmentation. (Right) A Pelger-Huët anomaly is visible here. This cell is a bilobular polymorphonuclear leukocyte that may occur on a congenital basis (without clinical significance) or may be acquired in myelodysplasia, in myeloproliferative diseases or with toxic stress on the bone marrow, as a result of treatment with chemotherapeutic drugs. (Oil immersion; × 1,000)
The myelodysplastic syndrome is subclassified into five types (Table 3). This subclassification has been of some value in following the natural history of myelodysplasia and in assessing the frequency of and times for conversion into acute leukemia. Refractory anemia with leukemia in transformation can transform to acute leukemia within months, whereas refractory anemia with excess blasts and chronic myelomonocytic leukemia may do so within two or three years. Refractory anemia and refractory anemia with sideroblasts may convert to leukemia within five or more years. Better estimations of life span and conversion to acute leukemia utilize the degree of cytopenia, cytogenetic abnormalities and percentage of leukemic cells in the marrow.10 Unfortunately, the type of leukemia associated with transformed myelodysplasia is acute nonlymphocytic leukemia (ANLL), which is resistant to therapy. This is in contrast to de novo ANLL, in which complete remission occurs in more than 50 percent of cases.
TABLE 3 Classification of Myelodysplastic Syndrome
| Refractory anemia |
| Refractory anemia with excess blasts |
| Refractory anemia with leukemia in transformation |
| Refractory anemia with sideroblasts |
| Chronic myelomonocytic leukemia |
In some patients, myelodysplastic syndrome occurs in association with preceding disorders such as malignancy and bone marrow dysfunction.11 In others, myelodysplasia may follow radiotherapy or chemotherapy. These secondary forms of myelodysplasia may also terminate in refractory ANLL. This outcome is one of the tragedies of modern medicine: an effective or curative treatment for a serious disease results in the development of a new disease that destroys life.
Compared with the clinical course of acute leukemia, the course of myelodysplasia is complicated more by anemia, infections and bleeding. Supportive care can be provided with transfusions, antibiotics or marrow growth stimulants, such as granulocyte colony–stimulating factor and erythropoietin.12,13 In some types of myelodysplastic syndrome, chemotherapy with topotecan (Hycamtin) has been tried with some success.14 However, no form of therapy prevents the conversion to acute leukemia except allogeneic bone marrow transplantation, an option that is available only in younger patients.15,16 Patients and family members need help with management of and adjustment to this chronic, irreversible disorder, which not only requires frequent medical attention but also can convert to a refractory and terminal acute leukemia.
