
Am Fam Physician. 1999;59(5):1190-1196
See related patient information handout on congenital adrenal hyperplasia, written by the authors of this article.
Congenital adrenal hyperplasia was once considered a rare inherited disorder with severe manifestations. Mild congenital adrenal hyperplasia, however, is common, affecting one in 100 to 1,000 persons in the United States and frequently eluding diagnosis. Both classic and nonclassic forms of the disease are caused by deficiencies in the adrenal enzymes that are used to synthesize glucocorticoids. The net result is increased production from the adrenal gland of cortisol precursors and androgens. Even mild congenital adrenal hyperplasia can result in life-threatening sinus or pulmonary infections, orthostatic syncope, shortened stature and severe acne. Women with mild congenital adrenal hyperplasia often present with hirsutism, oligomenorrhea or infertility. Congenital adrenal hyperplasia is diagnosed by demonstration of excess cortisol precursors in the serum during an adrenal corticotropic hormone challenge. Diagnosis of congenital adrenal hyerplasia in fetuses that are at risk for congenital adrenal hyperplasia can be determined using human leukocyte antigen haplotype or by demonstration of excess cortisol precursors in amniotic fluid. Treatment includes carefully monitored hormone replacement therapy. Recognition of the problem and timely replacement therapy can reduce morbidity and enhance quality of life in patients that are affected by congenital adrenal hyperplasia.
Classical congenital adrenal hyperplasia is rare, affecting only one in 14,000 patients, but mild forms of the disease may occur in one of every 100 to 1,000 persons.1,2 The condition is caused by a deficient synthesis of cortisol; most cases are related to 21-hydroxylase or 11-β hydroxylase deficiency3–5 (Figure 1). The affected enzyme can be totally or partially impaired. The degree of enzyme insufficiency determines the severity of the condition.2,5
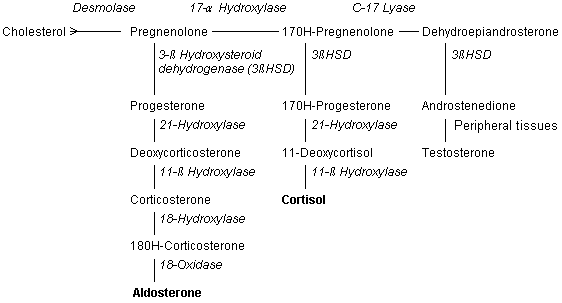
The hallmark of congenital adrenal hyperplasia is inadequate production of glucocorticoids.1 Patients with mild congenital adrenal hyperplasia are frequently unable to mount sufficient stress responses to trauma and infection. Glucocorticoid precursors accumulate in these persons and are converted to androgenic steroids, causing shortened stature, early puberty, severe acne, and virilization and infertility in females.2,3,5,6 Mineralocorticoid synthesis can also be affected, resulting in electrolyte disturbances, hypotension and syncope.5,6
Enzyme Pathways and Genetics
21-HYDROXYLASE
Ninety percent of patients with congenital adrenal hyperplasia have 21-hydroxylase deficiency.2–4,6 Because this enzyme functions in both glucocorticoid and mineralocorticoid synthesis, some patients with 21-hydroxylase deficiency have insufficient amounts of cortisone and aldosterone (Figure 2). These persons have the “salt-wasting” form of congenital adrenal hyperplasia, with hyponatremia, hypovolemia, hyperkalemia and hypotension.1–4,6 The enzyme 21-hydroxylase is a chromosome 6, human leukocyte antigen (HLA)–linked, cytochrome P450 enzyme that is found in the smooth endoplasmic reticulum. Its DNA sequence can be altered by at least nine mutations, many of which leave the enzyme impaired but not totally inactive.2,3,6

11-β HYDROXYLASE
Deficiency of 11-β hydroxylase is found in 8 to 9 percent of patients with congenital adrenal hyperplasia.2,5 Glucocorticoid synthesis remains impaired but, in this disorder, deoxycortisol accumulates (Figure 3). Deoxycortisol and its metabolites have mineralocorticoid properties and may cause hypertension when they accumulate.2,3,7 Thus, simple blood pressure measurements may help determine the underlying type of congenital adrenal hyperplasia. The enzyme 11-β hydroxylase is a chromosome 8, cytochrome P450 enzyme located in the mitochondria. Known gene abnormalities include insertions, deletions, missense/nonsense codons, and point mutations. Some of these abnormalities result in severe dysfunction of the enzyme while others result in only partial impairment.3–5
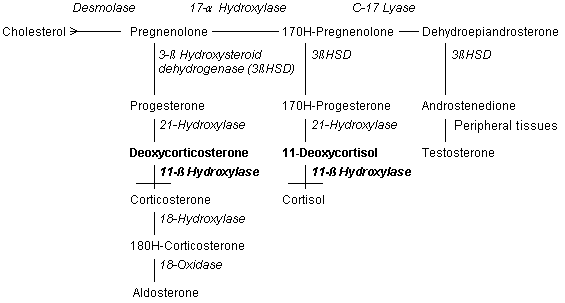
Classic 11-β hydroxylase deficiency occurs in approximately one per 100,000 births and occurs more frequently in Moroccan Jews. Mild congenital adrenal hyperplasia due to 11-β hydroxylase deficiency is more common, however, and may be responsible for 1 to 2 percent of cases of hirsutism and oligomenorrhea in women.3–5
Manifestations and Recognition
CLASSICAL CONGENITAL ADRENAL HYPERPLASIA
The classic form of congenital adrenal hyperplasia occurs when cortisol synthesis is extremely low. The disorder usually manifests in childhood. Hypersecretion of adrenal androgens causes masculinization of the external genitalia of the female fetus. Affected infants can have ambiguous genitalia or even erroneous gender assignment. Because testicles are not present to produce müllerian inhibiting factor, the internal female organs are intact.1,2,4
Children with classic congenital adrenal hyperplasia may lack sufficient amounts of cortisol to mount a stress response, and they frequently succumb to minor illnesses. Those who survive to adulthood experience premature puberty. Premature closure of the epiphyses results in short stature even though these children grow at an accelerated rate when young. Severe acne is also a frequent problem. Adult women with classic congenital adrenal hyperplasia may have pronounced hirsutism and amenorrhea.1–4,6
MILD CONGENITAL ADRENAL HYPERPLASIA
Mild congenital adrenal hyperplasia is much more common than the classic form.2,3,5,6 Men and women with mild congenital adrenal hyperplasia may have normal height compared with the general population, yet shortened stature when compared with their parents. Near-syncope may be a chronic or recurrent problem in these patients, and they frequently have a history of severe acne and mild hyperpigmentation. Some people with mild congenital adrenal hyperplasia can mount limited glucocorticoid stress responses and are thus never recognized as having the disorder. Others, however, have frequent illnesses and decompensate when challenged by common infections or minor trauma.1–4,6
Diagnosis and Treatment
ADULTS AND CHILDREN
When mild congenital adrenal hyperplasia is suspected, elevated serum levels of 17-hydroxyprogesterone suggest 21-hydroxylase deficiency, and elevated deoxycorticosterone/11-deoxycortisol levels suggest 11-β hydroxylase deficiency (Table 1; Figures 4 and 5). In patients who have few or no symptoms of mild congenital hyperplasia, the risks of treatment may outweigh the benefits. Patients who require treatment should be given glucocorticoid replacement therapy at the lowest dosage that achieves adrenal suppression; higher dosages can cause Cushingoid features and growth retardation.
| Children |
| Moderate to severe recurrent sinus or pulmonary infections |
| Severe acne |
| Hyperpigmentation, especially of the genitalia |
| Tall for age |
| Early onset of puberty |
| Adults |
| Childhood history as defined above |
| Syncope or near-syncope |
| Shortened stature compared with either parent |
| Hypotension (21-hydroxylase deficiency) |
| Hypertension (11-β hydroxylase deficiency) |
| Women |
| Clitorimegaly |
| Poorly developed labia |
| Hirsutism |
| Infertility |
| Polycystic ovary syndrome |
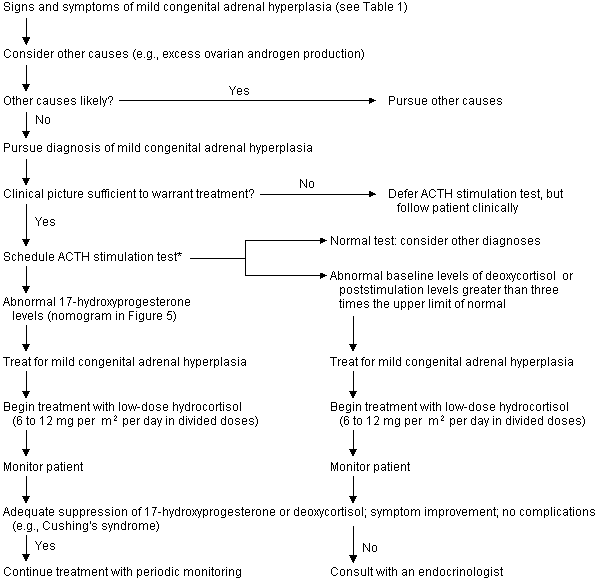
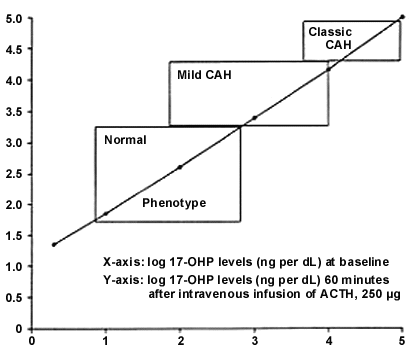
Maintenance therapy is generally achieved with hydrocortisone, in a dosage of 6 to 25 mg per m2 per day given in two to three divided doses.1,3,4,8 Hydrocortisone is preferred over other glucocorticoids because it is short acting and can be given in pulses that mimic natural cortisol secretion. Equivalent dosages of prednisone or dexamethasone can be used to simplify dosing regimens in noncompliant patients; however, hydrocortisone is more physiologically similar to cortisol and has a lower potential for growth suppression in children.3,4 Periods of physiologic stress, such as severe illness or surgery, require transient dosages of three to 10 times that used for maintenance therapy.1,3,4,8 Stress dosages are usually not needed in mild illnesses such as colds or otitis media.2–4
Corticosteroid replacement therapy must be approached carefully. Hydrocortisone dosages that return 17-hydroxyprogesterone/11-deoxycortisol levels to normal frequently induce Cushingoid features, whereas lower dosages may leave the effects of excess androgen production unchecked. Consultation with an endocrinologist is recommended for patients who require complex hormone regimens.
Many patients benefit from multidrug therapy. Even normotensive patients with 21-hydroxylase deficiency (Figure 2) may have improved adrenal suppression with the addition of the aldosterone analog fludrocortisone (Florinef) at dosages of 0.05 to 0.2 mg per day to their regimen.
The use of flutamide (Eulexin), an androgen inhibitor, in a dosage of approximately 10 mg per kg per day in three divided doses, in patients with all types of congenital adrenal hyperplasia may permit hydrocortisone to be given at lower dosages.8,9 Aromatase inhibitors that prevent conversion of androgens to estrogen (such as testolactone [Teslac], in a dosage of 40 mg per kg per day), may help children with mild congenital adrenal hyperplasia to achieve their height potential.1,8,9
PRENATAL THERAPY
If one child in a family is already affected by congenital adrenal hyperplasia, the HLA haplotypes of the parents and the affected child should be determined. Subsequent pregnancies can then be accurately evaluated for congenital adrenal hyperplasia by HLA haplotype analysis of chorionic villus or amniotic cells.2,3 In families at risk (e.g., one or both parents affected by some form of congenital adrenal hyperplasia) but with no affected children, congenital adrenal hyperplasia can be diagnosed during pregnancy by DNA analysis of chorionic villus or amniotic fluid cells or by measuring 17-hydroxy steroids in the amniotic fluid.10 All women with fetuses at risk for congenital adrenal hyperplasia should receive dexamethasone in a dosage of 0.02 mg per kg per day, divided into two or three daily doses.
Treatment should be initiated immediately on confirmation of pregnancy.4,10 Dexamethasone readily crosses the placenta and suppresses the fetal adrenal gland. If the fetus is male, dexamethasone therapy can be stopped until after the infant is born. Affected female fetuses, however, require treatment throughout pregnancy.
Treatment of affected fetuses with maternal dexamethasone reduces the incidence and severity of virilization of female fetuses. Birth weight, length, head circumference and congenital anomalies of affected infants whose mothers are treated with dexamethasone during pregnancy are comparable to those of infants who do not have congenital adrenal hyperplasia.10 Maternal complications from treatment with dexamethasone can be expected and include excess weight gain, mood swings and hypertension.10,11