Soft tissue sarcomas account for fewer than 1 percent of malignancies diagnosed annually in the United States. These tumors usually present as an asymptomatic mass. Any lesion larger than 5 cm in diameter should be considered suspicious. Radiographs should be obtained as the initial step in assessing a suspicious lesion. Magnetic resonance imaging has become the preferred diagnostic examination for tumors involving the extremities, and computed tomographic scanning may be the best technique for imaging lesions in the thoracic, abdominal, and head and neck areas. In general, the patient with a suspicious soft tissue mass located in a surgically difficult area should be referred to a regional center for biopsy and multidisciplinary consultation before resection is attempted. Careful preoperative planning is necessary for a good outcome. The prognosis for the patient with a soft tissue sarcoma is primarily determined by the grade, size and depth of the tumor and the presence of tumor at the surgical margins.
Although soft tissues constitute much of the human body, sarcomas account for fewer than 1 percent of all malignancies.1,2 In 1997, only 7,000 new cases of these tumors were diagnosed in the United States.2 Fewer than 20 percent of sarcomas are metastatic at the time of discovery, but the 4,300 deaths attributed to these masses in 1997 indicate how difficult they are to manage.3,4
Family physicians frequently encounter masses in their patients, but they will probably see only two soft tissue sarcomas over the course of a professional career. Preoperative diagnosis of these tumors improves the prognosis, and pretreatment planning is essential.5 The goal is to spare as much normal tissue as possible, with no local recurrence and no distant metastases. Limb- and tissue-sparing surgery and adjuvant radiation therapy have replaced amputation as the principal treatments for localized sarcomas.
Epidemiology
Soft tissue sarcomas arise from mesenchymal and neural crest tissue in the extremities, chest wall, retroperitoneum and mediastinum. Children are most likely to present with rhabdomyosarcomas or poorly differentiated sarcomas of the head and neck, or genitourinary area. In adults, soft tissue sarcomas occur most often in late middle age. Adults tend to have sarcomas of the extremities, retroperitoneum or head and neck. Most soft tissue sarcomas arise de novo, but a small percentage originate in injured tissues such as scars or radiation-exposed areas.6 In the rare inherited Li-Fraumeni syndrome, germ-line p53 mutations result in breast cancer, sarcomas and other neoplasms.7 Neurofibrosarcomas develop in 10 percent of patients with von Recklinghausen's disease.8
Little information is available on the prevalence of sarcomas in primary care practice. Fewer than one in 100 soft tissue masses seen by family physicians are found to be malignant, and only about one in three masses seen in cancer centers actually prove to be malignant.9–11
The only published population-based study on sarcomas is from the Southern Swedish Health Care Region.12 In this study, the reported annual incidence of 18 sarcomas per 1 million masses evaluated translates into one sarcoma seen every 15 to 20 years by a family physician with a panel of 2,500 patients. High-grade malignant fibrous histiocytoma, the soft tissue sarcoma most commonly seen in the study, occurred at a median age of 64 years. Metastasis occurred within three years in 40 percent of the patients with sarcomas.
The World Health Organization recognizes 121 types of soft tissue sarcoma13 (Table 1).10,11 Liposarcoma and malignant fibrous histiocytoma are the most common histopathologic types affecting the extremities and head and neck areas, whereas leiomyosarcomas are more common in the retroperitoneum and viscera. Many soft tissue sarcomas contain clonal chromosomal aberrations and dysfunctional RB1 or p53 suppressor genes.4,14
TABLE 1 Distribution of Diagnoses for Benign and Malignant Soft Tissue Tumors in a Large Referral Population*
| Malignant tumors (N = 12,370) | Incidence (%) | ||||
| Malignant fibrous histiocytoma | 24 | ||||
| Liposarcoma | 14 | ||||
| Undifferentiated sarcoma | 12 | ||||
| Leiomyosarcoma | 8 | ||||
| Malignant schwannoma | 6 | ||||
| Dermatofibrosarcoma protuberans | 6 | ||||
| Synovial sarcoma | 5 | ||||
| Fibrosarcoma | 5 | ||||
| Others | 20 | ||||
| Benign tumors (N = 18,677) | Incidence (%) | ||||
| Lipoma | 16 | ||||
| Fibrous histiocytoma | 13 | ||||
| Nodular fasciitis | 11 | ||||
| Hemangioma | 8 | ||||
| Fibromatosis | 7 | ||||
| Neurofibroma | 5 | ||||
| Schwannoma | 5 | ||||
| Others | 35 | ||||
*—Compiled from the computer-entered diagnoses of lesions seen at the Armed Forces Institute of Pathology from 1980 through 1989 and classified according to the system used by the World Health Organization.
Prognosis
With soft tissue sarcomas, the main determinants of prognosis are the grade, size and depth of the tumor, and the presence of tumor at the surgical margins3,15 (Table 2).9 Large high-grade tumors, characterized by poor cellular differentiation, are associated with a significantly higher incidence of local recurrence and distant metastasis16–18 (Table 3).15 Local spread tends to occur along fascial planes. Metastasis to the lung is common and is often the cause of death.4 Rhabdomyosarcomas, synovial sarcomas, and epithelioid and clear cell sarcomas of kidney frequently metastasize to lymph nodes.
Tumor size is an important determinant of survival. Patients with soft tissue sarcomas less than 5 cm in greatest diameter have a metastasis-free five-year survival rate of 81 percent15 regardless of tumor grade, depth and location, patient gender, surgical technique or use of adjuvant chemotherapy or radiation therapy.19 The five-year metastasis-free survival rate drops to 64 percent for tumors 5 to 9 cm in diameter and to 48 percent for tumors over 10 cm in diameter.15
Tumor depth is also a critical prognostic factor. If the tumor does not extend beyond the fascia, the five-year metastasis-free survival rate is 87 percent. In contrast, the five-year survival rate is 56 percent for deep tumors.15
A coordinated referral system can improve the prognosis for sarcomas. Regionalization of the Swedish health care system during the 1980s increased the likelihood that suspicious masses would be referred to cancer treatment centers. In these centers, patients with suspected sarcomas undergo imaging studies and needle biopsy before resection is attempted. This approach has lowered the incidence of local recurrence by two thirds. Swedish physicians repeatedly receive information to assist in identifying patients with suspicious tumors and accept that only one in every 10 referred patients will have malignant disease.12
Recognizing Soft Tissue Sarcomas
Soft tissue sarcomas usually present initially as an asymptomatic mass. Patients often wait an average of four months before seeking medical attention, and a definitive diagnosis may be delayed another six months in 20 percent of patients.20
Unfortunately, no one feature can reliably indicate if a mass is a sarcoma or whether it is a benign or malignant mass. Two thirds of sarcomas are deep seated and larger than most subcutaneous tumors.21 Compression of a nerve or blood vessel or interference with movement can cause symptoms. A vague but unrelated history of trauma often adds to the confusion.22 The physical examination reveals a firm, nontender mass that may seem well defined as a result of compression by surrounding tissue.
Diagnosis
Radiographs should be obtained as the first step in assessing a suspicious lesion, especially one that is over 5 cm in diameter. The radiologist should ascertain if the lesion arises from bone or is associated with a bony deformity. Calcifications are usually associated with benign disease, but they may also be present in liposarcomas or large malignancies with necrotic areas.23
Magnetic resonance imaging (MRI) has emerged as the preferred diagnostic modality for further study of extremity tumors after radiographs have been obtained (Figures 1 and 2). Compared with MRI studies, computed tomographic (CT) scans may provide more information about thoracic, head and neck, and abdominal tumors (Figure 3). MRI and CT scans differentiate benign from malignant lesions in 85 percent of patients.23–26 Ultrasonography can differentiate solid and cystic tumors, but it may confuse necrotic tumors with benign cysts.23,27
FIGURE 1.
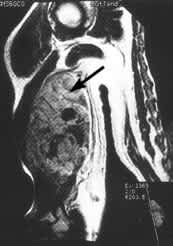
T9-weighted magnetic resonance image obtained with contrast medium. This coronal view of the right upper arm shows a pleomorphic liposarcoma (arrow).
FIGURE 2.
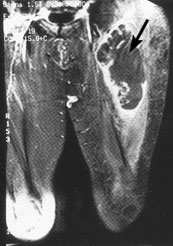
T1-weighted magnetic resonance image in which fat has been suppressed with contrast medium. This coronal view of the left leg shows a pleomorphic malignant fibrous histiocytoma (arrow). The tumor demonstrates necrosis after radiation and chemotherapy.
FIGURE 3.
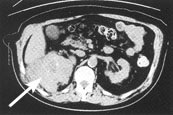
Computed tomographic scan obtained without contrast medium. The cross-sectional abdominal scan shows a retroperitoneal malignant fibrous histiocytoma (arrow).
A complete physical examination is indicated in all patients with suspicious masses. Most patients should undergo chest radiography and possibly CT scanning of the chest before biopsy samples are obtained to search for possible metastatic disease. Because of the high rate of residual tumor after unplanned excisions, a strategy of “excise and wait for the pathology report” should be avoided unless a mass is less than 5 cm in diameter and imaging studies indicate that the lesion is benign.5,28
Biopsy samples should always be obtained from suspicious masses. Where the biopsy procedure is performed—and by whom—is an important primary care decision. Sarcomas are rare, their histologic interpretation can be difficult, and biopsy procedures often procure only small amounts of tissue for study.29 The error rate for biopsies performed by orthopedic surgeons in community hospitals has been higher than the error rate for biopsy and interpretation procedures performed at cancer centers.30
Core-needle biopsy is the method of choice. Although fine-needle aspiration biopsy is somewhat easier to perform, it provides an inadequate specimen 18 percent of the time.31,32 In contrast, core-needle biopsy provides an adequate sample 93 percent of the time and correct differentiation of malignant and benign lesions more than 90 percent of the time. In addition, core-needle samples usually provide enough material to determine tumor grade.31,33
Treatment
With soft tissue sarcomas, the initial surgical procedure is extremely important. Both high- and low-grade tumors less than 5 cm in diameter may be treated by wide local excision (margins of at least 1 cm). The surgical margins must be carefully examined for residual tumor. Even at referral centers, 7 percent of patients require a second resection to achieve tumor-free margins.20
Outcomes for wide-margin excision of tumors less than 5 cm in diameter are equivalent whether or not adjuvant radiation therapy is employed. In contrast, high-grade tumors larger than 5 cm in diameter require wide surgical excision plus radiation therapy administered either preoperatively or postoperatively.3,20 Large tumors close to vascular, bony or nerve structures may be resected more successfully when radiation therapy is used preoperatively. Although radiation therapy decreases local recurrence dramatically, it may not affect survival because high-grade tumors tend to metastasize early.34
The role of chemotherapy in the treatment of soft tissue sarcomas remains unclear. Nonetheless, patients with high-risk tumors should be considered for entry into chemotherapy trials.15
Unfortunately, the 20 percent five-year survival rate for patients with lung metastases has yet to be improved.20
When to Refer
Because the initial surgical procedure is so important, the family physician creates greater opportunity for the patient when a soft tissue sarcoma is diagnosed before treatment. Referral to a cancer treatment center and an experienced cancer surgeon should be considered for any patient with a suspicious mass.
Preoperative diagnosis potentiates proper staging and the use of a multidisciplinary team (medical and surgical oncologists, radiation therapists, physiatrists and pathologists) to plan the best treatment. A “low suspicion” mass in a surgically accessible area requires less advance planning than a suspicious tumor in a technically difficult location. High-grade lesions require thoughtful management, regardless of location.
Treatment goals, including local tumor control, preservation of function and avoidance of amputation, should be discussed with the patient.
