Anemia is a common problem that is often discovered on routine laboratory tests. Its prevalence increases with age, reaching 44 percent in men older than 85 years. Normocytic anemia is the most frequently encountered type of anemia. Anemia of chronic disease, the most common normocytic anemia, is found in 6 percent of adult patients hospitalized by family physicians. The goals of evaluation and management are to make an accurate and efficient diagnosis, avoid unnecessary testing, correct underlying treatable causes and ameliorate symptoms when necessary. The evaluation begins with a thorough history and a careful physical examination. Basic diagnostic studies include the red blood cell distribution width, corrected reticulocyte index and peripheral blood smear; further testing is guided by the results of these studies. Treatment should be directed at correcting the underlying cause of the anemia. A recent advance in treatment is the use of recombinant human erythropoietin.
Anemia is defined as a decrease in the circulating red blood cell mass to below age-specific and gender-specific limits. In normocytic anemias, the mean corpuscular volume (MCV) is within defined normal limits, but the hemoglobin and hematocrit are decreased. The MCV is also age-specific (Figure 1),1 with normal values ranging from 70 femtoliter (fL) at one year of age to 80 fL at seven years and older.2
Most patients with anemia are asymptomatic. Therefore, the condition is most often discovered by laboratory evaluation, usually on routine testing as part of the general physical examination or for reasons other than suspected anemia. Anemia should be considered a sign, not a disease.3 It can be caused by a variety of systemic disorders and diseases, as well as primary hematologic disorders.
Approximately 4.7 million Americans have anemia.4 Population-based estimates indicate that this condition affects 6.6 percent of males and 12.4 percent of females. The prevalence of anemia increases with age and is 44.4 percent in men 85 years and older.5 Although the elderly are more prone to develop anemia, older age is not of itself a cause of the condition.6
Etiology
Normocytic anemias may be thought of as representing any of the following: a decreased production of normal-sized red blood cells (e.g., anemia of chronic disease, aplastic anemia); an increased destruction or loss of red blood cells (e.g., hemolysis, posthemorrhagic anemia); an uncompensated increase in plasma volume (e.g., pregnancy, fluid overload); or a mixture of conditions producing microcytic and macrocytic anemias.
It should be noted that in the initial stage, nearly all anemias are normocytic. The major primary causes of normocytic anemia are given in Table 1.
TABLE 1 Primary Causes of Normocytic Anemias*
| Increased red blood cell loss or destruction | |||||
| Acute blood loss | |||||
| Hypersplenism | |||||
| Hemolytic disorders | |||||
| Congenital conditions | |||||
| Hemoglobinopathies | |||||
| Homozygous sickle cell disease (hemoglobin SS disease) | |||||
| Heterozygous sickle hemoglobin C disease (hemoglobin SC disease) | |||||
| Disorders of red blood cell membranes | |||||
| Hereditary spherocytosis | |||||
| Hereditary elliptocytosis | |||||
| Red blood cell enzyme deficiencies | |||||
| Glucose-6-phosphate dehydrogenase deficiency | |||||
| Pyruvate kinase deficiency | |||||
| Acquired conditions | |||||
| Mechanical hemolysis | |||||
| Macrovascular disorders | |||||
| Microangiopathic disorders | |||||
| Disseminated intravascular coagulopathy | |||||
| Hemolytic-uremic syndrome | |||||
| Thrombotic thrombocytopenic purpura | |||||
| Autoimmune hemolytic anemias | |||||
| Warm-reactive anemias | |||||
| Cold-reactive anemias | |||||
| Drug-induced anemias | |||||
| Paroxysmal nocturnal hemoglobinuria | |||||
| Decreased red blood cell production | |||||
| Primary causes | |||||
| Marrow hypoplasia or aplasia | |||||
| Myelopathies | |||||
| Myeloproliferative diseases | |||||
| Pure red blood cell aplasia | |||||
| Secondary causes | |||||
| Chronic renal failure | |||||
| Liver disease | |||||
| Endocrine deficiency states | |||||
| Anemia of chronic disease | |||||
| Sideroblastic anemias | |||||
| Overexpansion of plasma volume | |||||
| Pregnancy | |||||
| Overhydration | |||||
*—Mean corpuscular volume of 81 to 99 fL.
Decreased Red Blood Cell Production
ANEMIA OF CHRONIC DISEASE
Anemia of chronic disease is the most common normocytic anemia and the second most common form of anemia worldwide (after iron deficiency anemia).7 The MCV may be low in some patients with this type of anemia. The pathogenesis of anemia of chronic disease is multifactorial and is related to hypo-activity of the bone marrow, with relatively inadequate production of erythropoietin or a poor response to erythropoietin, as well as slightly shortened red blood cell survival.
Anemia of chronic disease is associated with a wide variety of chronic disorders, including inflammatory conditions, infections, neoplasms and various systemic diseases. The diagnosis of anemia of chronic disease is not usually applied to the anemias associated with renal, hepatic or endocrine disorders. Patients with these disorders may not display the hallmark ferrokinetic profile of anemia of chronic disease (i.e., decreased serum iron level, decreased transferrin level, or normal or elevated ferritin levels, all of which result in iron being present but inaccessible for use).3,8–10
ENDOCRINE DEFICIENCY
Endocrine deficiency states, including hypothyroidism, adrenal or pituitary insufficiency, and hypogonadism, may cause secondary bone marrow failure because of reduced stimulation of erythropoietin secretion. Hyperthyroidism may also cause normocytic anemia.3,9
RENAL FAILURE
Anemia occurs in acute and chronic renal failure. The anemia is usually normocytic but may be microcytic. In renal failure, anemia occurs in part because uremic metabolites decrease the lifespan of circulating red blood cells and reduce erythropoiesis.
Anemia secondary to uremia is characterized by inappropriately low erythropoietin levels, in contrast to the normal or high levels that occur with most other causes of anemia. To further confuse the presentation, serum iron levels and the percentage of iron saturation are often low, apparently because of negative acute-phase reactions.10 Furthermore, the serum creatinine level and the degree of anemia may not correlate well.3
OTHER CAUSES
Other causes of decreased red blood cell production include bone marrow infiltration, fibrosis, various myeloproliferative diseases and sideroblastic anemias. These uncommon disorders are generally diagnosed by bone marrow biopsy.
Increased Red Blood Cell Destruction or Loss
HEMOLYTIC ANEMIAS
Hemolytic anemias other than the alloimmune hemolytic anemias of newborns (e.g., Rh or ABO incompatibility) can be categorized as congenital or acquired (Table 2).3,9,11–13
TABLE 2 Selected Causes of Hemolytic Anemias
| Disorder | Most common clinical features | Features of peripheral blood smear | Laboratory tests | |
|---|---|---|---|---|
| Congenital conditions | ||||
| Homozygous sickle cell disease (hemoglobin SS disease) | Vaso-occlusive crises, splenomegaly, cerebrovascular accidents, priapism, hand-foot syndrome, acute chest syndrome | Sickle cells | Hemoglobin electrophoresis | |
| Heterozygous sickle hemoglobin C disease (hemoglobin SC disease) | Generally similar to homozygous sickle cell disease, except associated with fewer infections, less hemolysis and fewer crises, but more retinopathy and aseptic necrosis | Sickle cells, target cells | Hemoglobin electrophoresis | |
| Hereditary spherocytosis | Childhood anemia, splenomegaly, jaundice | Spherocytes | Osmotic fragility test | |
| Hereditary elliptocytosis | Variable: asymptomatic carrier state to severe hemolysis | Elliptocytes | 25 percent or more of red blood cells elliptocytic on peripheral blood smear | |
| G6PD deficiency | Transient hemolysis following exposure to oxidative drug | Normal | G6PD activity | |
| Pyruvate kinase deficiency | Variable: severe anemias in newborns to no symptoms in adults | Normal | Red blood cell P-50 level (screening); red blood cell pyruvate kinase activity (confirmatory) | |
| Acquired conditions | ||||
| Microangiopathic disorders | Thrombocytopenia, schistocytes | |||
| Disseminated intravascular coagulopathy | Bleeding and/or intravascular hemolysis | Hypofibrinogenemia; increases in partial thromboplastin time, prothrombin time, fibrin split products and thrombin time | ||
| Hemolytic-uremic syndrome | Fever, jaundice, bleeding, central nervous system changes, renal failure; generally occurs in children | Increased creatinine level | ||
| Thrombotic thrombocytopenic purpura | Purpura, fever, central nervous system changes; generally occurs in adults | |||
| Mechanical hemolysis | Mild to moderate anemia; frequently, iron deficiency, second-degree chronic urinary loss; history of heart valve replacement or valvular disease | Schistocytes | None | |
| Paroxysmal nocturnal hemoglobinuria | Recurrent abdominal pain, vomiting, headache, eye pain; venous thromboses; leads to iron deficiency anemia | Normal | Sucrose hemolysis (screening); Ham's test (confirmatory) | |
G6PD = glucose-6-phosphate dehydrogenase; P-50 = oxygen half-saturation pressure of oxygen.
Congenital hemolytic anemias include the hemoglobinopathies (homozygous sickle cell disease [hemoglobin SS disease], heterozygous sickle hemoglobin C disease [hemoglobin SC disease]), red blood cell membrane disorders and red blood cell enzyme deficiencies.11,12
Homozygous sickle cell disease is the most common cause of hemolytic normocytic anemias in children. Because of longevity, this disease is also becoming an increasingly prevalent cause of these anemias in adults.11–13
Hereditary spherocytosis is the most common red blood cell membrane disorder. It usually presents in childhood with anemia, jaundice and splenomegaly. Pigment gallstones, delayed growth and dysmorphic features may occur. Hereditary elliptocytosis ranges from an asymptomatic carrier state to severe hemolytic anemia.11–13
Red blood cell enzyme deficiencies include glucose-6-phosphate dehydrogenase (G6PD) and pyruvate kinase deficiencies. More than 300 varieties of G6PD deficiency have been identified. The southern Mediterranean variety, referred to as “favism,” is best known, but the most common variant in the United States is a less severe X-linked disorder that affects 10 percent of black males. Persons with the U.S. variant may experience an acute, self-limited hemolytic episode after exposure to causes of oxidative stress, including sulfa drugs, nitrofurantoin (Furadantin), phenazopyridine (Pyridium) and antimalarial drugs.11,12
Acquired hemolytic anemias include autoimmune hemolytic anemias, mechanical hemolysis and paroxysmal nocturnal hemoglobinuria.12 Autoimmune hemolytic anemias primarily occur in persons older than 40 years. The most common and typically most severe of these anemias are those caused by warm-reactive antibodies. Autoimmune hemolytic anemias caused by cold-reactive antibodies most commonly follow Mycoplasma pneumonia or infectious mononucleosis.
Drugs that induce autoimmune hemolytic anemias include methyldopa (Aldomet), penicillins, cephalosporins, erythromycin, acetaminophen (e.g., Tylenol) and procainamide (Pronestyl).
Paroxysmal nocturnal hemoglobinuria generally presents as a chronic hemolytic anemia. Classic nocturnal hemoglobinuria is seldom seen.12
UNCOMPENSATED BLOOD LOSS
Acute posthemorrhagic anemia occurs with gastrointestinal bleeding, bleeding from an external wound or, less obviously, retroperitoneal bleeding or bleeding into a hip fracture. A healthy young person would be expected to tolerate rapid loss of 500 to 1,000 mL of blood (10 to 20 percent of the total blood volume) with few or no symptoms, although about 5 percent of the general population would have a vasovagal reaction.14 Indeed, healthy young persons at rest may tolerate an acute isovolemic reduction of hemoglobin volume to a level of 5 g per dL (50 g per L) without impairment of critical oxygen delivery.15
HYPERSPLENISM
Hypersplenism leads to anemia only after the spleen reaches three to four times its normal size, as may occur in cirrhosis, chronic infections and myeloproliferative diseases. The anemia is primarily caused by the removal of red blood cells from the circulation, but increased destruction of red blood cells is usually a contributing factor.16
Normocytic Anemia in Children
The prevalence of anemias caused by iron deficiency or lead toxicity continues to decline in the United States.17 As a result, normocytic anemias are constituting a larger proportion of cases in the pediatric age group.
Iron deficiency, which in its early stages is usually characterized by a normal MCV, is still a common cause of mild normocytic anemia in children beyond the neonatal period. Other common childhood normocytic anemias are the result of acute bleeding, sickle cell anemia, red blood cell membrane disorders and current or recent infections (particularly in younger children).2,17 Aplastic crises in patients of any age who have chronic hemolytic anemias are frequently precipitated by human parvovirus B19 infection.2,12,13,18
Most anemias in children can be diagnosed with a basic work-up that includes a complete blood cell count (CBC), a corrected reticulocyte index, a peripheral blood smear and targeted studies of the peripheral blood (e.g., hemoglobin electrophoresis).
Although bone marrow examinations are generally unnecessary, one study found that when the basic laboratory studies and historical and physical evidence were unrevealing, bone marrow specimens yielded a specific diagnosis in 92 percent of children.18 The most frequent diagnosis in this study was transient erythroblastopenia of childhood, a common, generally mild, self-limited red blood cell aplasia of unknown etiology. This entity must be distinguished from Blackfan-Diamond syndrome, a rare, usually macrocytic and probably genetic disorder of infants. Blackfan-Diamond syndrome is a congenital erythroid hypoplasia that usually does not spontaneously remit.3,9
Diagnosis
Physicians are sometimes inefficient in their evaluation of normocytic anemia, either ordering an excessive battery of tests or foregoing testing entirely in the belief that a cause is not likely to be found.19 The first step in the evaluation of anemia is to correlate the finding of anemia with the information obtained from the patient's history and physical examination. In many instances, this approach allows a working diagnosis to be made and many disorders to be eliminated.
Most published algorithms for the diagnosis of normocytic anemia begin with an examination of the peripheral blood smear20 or a corrected reticulocyte index.2,9,21 The red blood cell distribution width is a measure of the variability of the size (anisocytosis) of the cells and is usually reported as a component of automated CBCs. Therefore, a practical and useful first step is to use the red blood cell distribution width to help categorize the normocytic anemia as heterogeneous (e.g., hemolytic anemia) or homogeneous (e.g., anemia of chronic disease).2 In patients with a mild homogeneous normocytic anemia (hematocrit of 30 percent or greater) and a known chronic disease, anemia of chronic disease is highly likely, and bone marrow biopsy may not be necessary (Figure 2).21
FIGURE 2. Evaluation of Normocytic Anemia
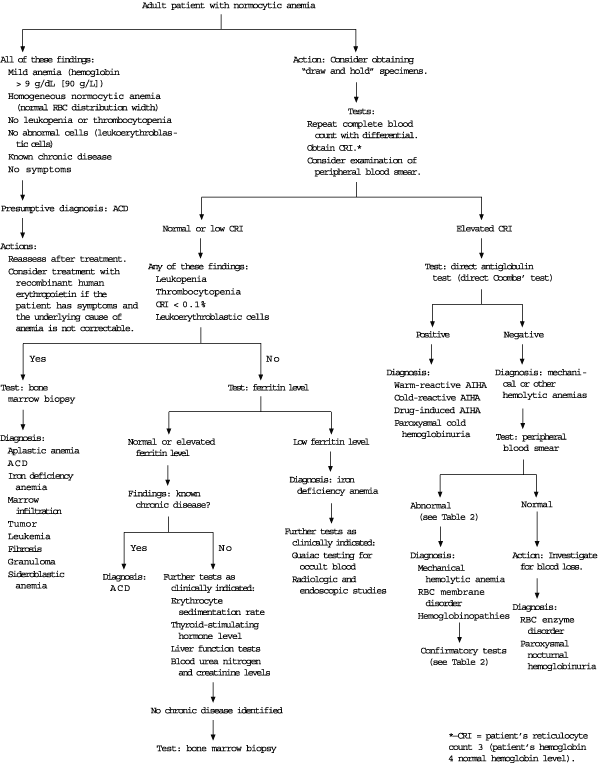
Approach to the evaluation of normocytic anemia in adults. (RBC = red blood cell; CRI = corrected reticulocyte index; ACD = anemia of chronic disease; AIHA = autoimmune hemolytic anemia)
Adapted with permission from Brown RG. Normocytic and macrocytic anemias. Postgrad Med 1991;89(8):125–32,135–6.
‘DRAW AND HOLD’ STRATEGY
Because the diagnosis of normocytic anemia usually proceeds in a step-wise fashion that begins with the corrected reticulocyte index and examination of the peripheral blood smear, a patient-friendly, cost-effective and time-efficient strategy is to use a “draw and hold” order for possible later testing. Most laboratories do not charge to hold tubes, and tests can usually be added up to one week after specimens are obtained. The physician should check with the local laboratory to determine the number and type of specimens that need to be obtained.
PERIPHERAL BLOOD SMEAR
The examination of the peripheral blood smear often yields diagnostic clues or confirmatory evidence. Easily recognized red blood cell findings related to normocytic anemias include the following: large polychromatic “shift cells,” which represent reticulocytosis; target cells, which may be found in liver disease; basophilic stippling, which may be present in hemolytic anemias; and mixtures of large and small red blood cells, which may suggest the presence of mixed microcytic and macrocytic disease processes (a finding that should be suggested by an elevated red blood cell distribution width).
Other findings include burr cells (uremia), spherocytes (hereditary spherocytosis, autoimmune hemolysis, G6PD deficiency), elliptocytes (hereditary elliptocytosis), schistocytes (microangiopathic processes), bite or blister cells (where all of the hemoglobin appears to be pushed to one side of the cell, G6PD deficiency) and nucleated red blood cells (hemolytic anemia, acute blood loss). These findings may be present in other anemias and in other conditions.3,9,10
The corrected reticulocyte index, along with the white blood cell and platelet counts, indicates whether the bone marrow is functioning appropriately. The corrected reticulocyte index should be elevated in patients with an acute anemia but a competent bone marrow.
ILLUSTRATIVE CASES
Case 1. A 50-year-old woman who had been taking aspirin for a flare of rheumatoid arthritis presented with mild epigastric pain. A CBC was ordered, and a guaiac test for occult blood was performed. The guaiac test was negative.
The CBC revealed a normocytic anemia (hemoglobin count, 11 per mm3 [11 × 106 per L]; hematocrit, 33 percent [0.33]; MCV, 84 fL), with a red blood cell distribution width of 41 fL (normal range: 39 to 47 fL). A reticulocyte count and “draw and hold” specimens were ordered. The corrected reticulocyte index was 1.0 percent.
Ferritin and serum iron levels were obtained from the stored specimens. These tests revealed an elevated ferritin level and a low serum iron level, findings consistent with a diagnosis of anemia of chronic disease related to the patient's rheumatoid arthritis.
Case 2. A 44-year-old woman presented with the complaint of fatigue. Her physical examination was unremarkable.
A CBC revealed normocytic anemia (hemoglobin count, 11 per mm3 [11 × 106 per L]; hematocrit, 33 percent [0.33]; MCV, 84 fL), with an elevated red blood cell distribution width of 53 fL. A reticulocyte count and “draw and hold” specimens were ordered. The corrected reticulocyte index was elevated (3.6 percent).
Examination of a peripheral blood smear from the stored specimens was normal. A direct antiglobulin test (direct Coombs' test) was positive, and a preliminary diagnosis of autoimmune hemolytic anemia was made.
Treatment
The treatment of a normocytic anemia begins with timely identification of its cause. In most patients, therapy is individualized to the underlying disorder. Treatments may include avoidance of trigger exposure in patients with hemolytic anemia, correction of iron, folate or vitamin B12 deficiency in patients with mixed disorders, or splenectomy in patients with hypersplenism.12,13
Anemia of renal disease is associated with a relative underproduction of erythropoietin, and inappropriate erythropoietin levels appear to contribute significantly to anemia of chronic disease. With the development of recombinant human erythropoietin (r-HuEPO; epoetin alfa [Epogen]), there has been considerable interest in finding out whether exogenous erythropoietin administration would improve anemia.
The effects of r-HuEPO administration have been studied in a variety of disorders. In a trial conducted in 1990,22 all 11 patients with anemia related to rheumatoid arthritis reached a normal hematocrit after 24 weeks. Since then, r-HuEPO has been tested in patients with anemia of chronic disease secondary to acquired immunodeficiency syndrome, malignancy, inflammatory bowel disease, renal disease and other disorders.23,24 Quality-of-life parameters in responders improved significantly.
Therapy with r-HuEPO is very expensive and should never replace treatment of the underlying cause of an anemia. R-HuEPO is an indicated therapy for anemia of renal disease. In this situation, its use should be based on clinical and quality-of-life issues rather than specific hemoglobin levels.10 There are no consistent guidelines for r-HuEPO therapy in patients with anemia of chronic disease, although response rates of 40 to 80 percent may be achieved.8
Erythropoietin also appears to be useful prophylactically in patients undergoing autologous blood donation and certain surgical procedures.25
In all patients, treatment of anemia should include the provision of optimal nutrition and supportive care.
