Although most annular lesions will be typical of a dermatophytosis, physicians must consider other possible diagnoses. Tinea corporis can often be diagnosed on the basis of a positive potassium hydroxide examination. Topical and systemic antifungals are usually curative. Pityriasis rosea is characterized by small, fawn-colored lesions distributed along skin cleavage lines. Treatment is symptomatic. Granuloma annulare is characterized by nonscaly, annular plaques with indurated borders, typically on the extremities. One half of cases resolve spontaneously within two years. Sarcoidosis can present as annular, indurated plaques similar in appearance to the lesions of granuloma annulare. Diagnosis is based on histopathology and the involvement of other organ systems. Hansen's disease can mimic tinea corporis by presenting as one or more annular, sometimes scaly, plaques. Urticaria may affect 10 to 20 percent of the population. The annular plaques lack scale and are evanescent. Subacute cutaneous lupus erythematosus can present in an annular form on sunexposed surfaces or in a papulosquamous form. Erythema annulare centrifugum typically presents as annular patches with trailing scale inside erythematous borders.
Annular lesions are extremely common and striking in appearance but can also be misleading. The term “annular” stems from the Latin word “annulus,” meaning ringed.1 The lesions appear as circular or ovoid macules or patches with an erythematous periphery and central clearing. The most common cause of annular lesions encountered in the adult population is dermatophytosis, which may be successfully diagnosed without a biopsy. However, other conditions may present with much the same appearance (Table 1). The clinician must exclude other diagnoses, especially if the patient has failed previous treatment for dermatophytosis.
Tinea Corporis
Tinea corporis refers to a dermatophytosis or superficial fungal infection of the skin, other than on the hands, feet, scalp, face or groin.2 This condition is also commonly referred to as ringworm, a misnomer that stems from the annular appearance once believed to be caused by invasive worms. The main causes belong to three genera: Trichophyton, Microsporum and Epidermophyton. The most common cause of tinea corporis in the United States is infection with Trichophyton rubrum, Trichophyton tonsurans, Trichophyton mentagrophytes and Microsporum canis.1 All dermatophytes are aerobic, and they share the ability to assimilate keratin, thus allowing them to penetrate the keratinized layers of the stratum corneum.3,4
Humans may become infected with tinea corporis through close contact with infected persons, animals or soil.4 Occasionally, autoinoculation from infected nails, scalp or feet may occur.1 The peak incidence of infection is after puberty, although patients may present in the preadolescent period.4 There is no sexual predilection.3 Climate and personal habits are the main determinants of these infections.3 Fungal infections are facilitated by warm, moist environments (such as public showers and swimming pools) and the sharing of towels, clothing and toiletries. Prolonged use of systemic corticosteroids may render a person more vulnerable to fungal infections such as this.4
Patients present with well-demarcated, erythematous papules or plaques on the skin.2 Patients may note that these lesions have gradually enlarged over time.1 The borders represent the active edge of the lesion and may be raised or scaly3 (Figure 1).
KOH PREPARATION
An accurate diagnosis requires demonstration of the fungus using a potassium hydroxide (KOH) microscopic slide preparation. Using a scalpel blade, the scales are scraped at the active border of the lesion, with particular care not to cause pain or bleeding. The specimen is transferred to a glass slide, and a coverslip is placed on top to protect the specimen. A drop of 10 to 15 percent KOH, with or without dimethyl sulfoxide (DMSO; Rimso-50), is added. The specimen is then gently heated; specimens prepared with DMSO do not require heating. Overheating or boiling the specimen may cause the KOH to crystallize, which leads to artifacts.
First, the specimen should be examined under low-power magnification. The KOH and the heat dissolve the keratinocyte cell membrane, leaving behind easily visualized septate hyphae, which are long and may be straight or wavy. These structures may also branch and generally have a uniform diameter. If repeated KOH preparations are negative in a patient with a clinically suspected dermatophyte infection, fungal cultures are recommended. The cultures may take two to four weeks for growth.
TREATMENT OF TINEA CORPORIS
Although treatment of tinea corporis can be relatively simple, patients must prevent reinfection by treating or avoiding contaminated soil, animals and people. Numerous topical and systemic antifungal agents are available.1 It is usually best to attempt topical treatment first. Topical imidazoles, such as clotrimazole (Lotrimin) and miconazole (Monistat), and allylamines, such as naftifine (Naftin) terbinafine (Lamisil) and butenafine (Mentax), are generally effective in treating localized lesions.1,4
The use of systemic antifungal agents may be considered in patients with the following conditions: tinea corporis resistant to or intolerant of topical therapy; disabling or extensive disease; chronic infection; primary or secondary immunosuppression; and dermatophytosis of hyperkeratotic areas such as the palms and the soles.4 Among the systemic antifungal agents available, griseofulvin (Grisactin), terbinafine and itraconazole (Sporanox) are effective.1 Although ketoconazole (Nizoral) is effective, it has potentially dangerous side effects, such as hepatic injury and sterol inhibition.1,2 The usual course of oral therapy for tinea corporis is two weeks. Unfortunately, systemic antifungal agents can be expensive and have some drug-to-drug interactions.1
TABLE 1 Comparison of Annular Lesions
| Diagnosis | Clinical presentation | Treatment options |
|---|---|---|
| Tinea corporis | Scaly, annular, erythematous plaques or papules on glabrous skin | Topical and systemic antifungals |
| Pityriasis rosea | Small, fawn-colored, oval patches with fine scale along the borders, following skin cleavage lines | Topical and systemic corticosteroids; UVA, UVB |
| Granuloma annulare | Indurated, nonscaly, skin-colored annular plaques and papules, usually on the extremities | Topical and intralesional corticosteroids |
| Sarcoidosis | Indurated, erythematous plaques | Topical, intralesional and systemic corticosteroids; antimalarials; thalidomide |
| Hansen's disease | Erythematous annular plaques, with or without scale | Dapsone; rifampin (Rifadin) |
| Urticaria | Evanescent annular, nonscaly, erythematous plaques | Oral antihistamines |
| Subacute cutaneous lupus erythematosus | Annular or papulosquamous plaques, with or without scale, on sun-exposed areas | Topical, intralesional and systemic corticosteroids; antimalarials |
| Erythema annulare centrifugum | Annular patches with trailing scale inside erythematous borders | Topical and systemic corticosteroids; oral antihistamines; treatment of the underlying cause |
UVA = ultraviolet A light; UVB = ultraviolet B light.
Pityriasis Rosea
Pityriasis rosea is a common, self-limited papulosquamous eruption that may resemble tinea corporis.5,6 Although its etiology is unknown, current data suggest that pityriasis rosea may be a viral exanthem.6,7 It usually occurs during the second to fourth decades of life,6,8 with peak incidence in teenagers, adolescents and young adults.7 Women are more likely to be affected, but the exact rate is unknown, and there is no racial predilection.6 Pityriasis rosea does appear to occur more often during the spring and autumn seasons.5–7
FIGURE 1.
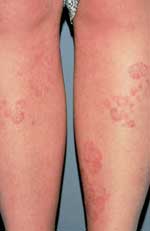
Tinea corporis—well-demarcated, erythematous plaques with raised, scaly borders.
Although most pityriasis rosea lesions are macules, papules or plaques, the initial presenting lesion, also known as the “herald patch,” may appear as an annular lesion with an erythematous, raised border, fine scale and central clearing6 (Figure 2). The herald patch is generally a singular, ovoid macule located on the trunk and can range from 2 to 10 cm in diameter.5–8 Although most patients are asymptomatic except for the rash,7 5 percent of patients complain of headaches, malaise, arthralgias, chills, nervousness, vomiting, diarrhea or constipation before the appearance of the herald patch.7–8 Within seven to 14 days, patients develop an eruption characterized by lesions reminiscent of the herald patch.1,6 The rash consists of small, fawn-colored, oval macules, which exhibit peripheral, scaly collarettes similar to those on the herald patch. These lesions are generally bilaterally symmetric and may be located anywhere on the body, especially on the neck, trunk and proximal extremities. Because the lesions follow the skin cleavage lines, they have the characteristic appearance of a Christmas tree.1,6–8
FIGURE 2.
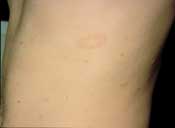
Pityriasis rosea—fawn-colored patch with fine scale along the border.
There is no effective treatment for pityriasis rosea, except that patients who experience severe pruritus may benefit from topical corticosteroids and oral antihistamines.6 Severe cases can be treated with ultraviolet A (UVA) light, ultraviolet B (UVB) light or systemic corticosteroids. Spontaneous resolution occurs within six to eight weeks, during which time new crops of lesions continue to appear while others progressively fade.6–8 Fewer than 3 percent of patients suffer recurrences.8 Patients thought to have pityriasis rosea should be screened for syphilis, because the two eruptions often have a similar appearance.
Granuloma Annulare
Granuloma annulare is an idiopathic, selflimited cutaneous condition that is common in adults and children. The condition is considered benign and is characterized by smooth, skin-colored annular plaques and papules.1 The lesions are usually found on the hands, feet, wrists and ankles but can potentially occur anywhere on the body.9 Although the condition is generally asymptomatic, some patients may note mild pruritus.9 Granuloma annulare has a higher incidence in women,9 and it develops before the fourth decade of life in the majority of patients.1
Granuloma annulare can be categorized as localized, generalized, perforating, subcutaneous1,9 and actinic (granuloma annulare on sun-exposed skin).
The most common clinical form is localized granuloma annulare, which accounts for approximately 75 percent of all cases.1 Patients generally present with one or several skin-colored to erythematous papules.1,9 The lesions have smooth, raised borders1 and range from 1 to 5 cm in diameter.1,9 They may be isolated or coalesce into plaques (Figure 3). The most commonly affected site is the legs, with the palms, scalp and plantar surfaces usually being spared.9 Unlike tinea corporis, the lesions of granuloma annulare have no scaling or associated vesicles or pustules.1
Roughly one half of patients with localized granuloma annulare experience spontaneous resolution within two years.9 Patients with generalized granuloma annulare have more widespread involvement, usually with 10 or more lesions.1 Spontaneous resolution is less common in these patients than in patients with localized granuloma annulare, and remission in fewer than three to four years is unlikely.9
Perforating granuloma annulare lesions are small, umbilicated papules that are found predominantly on the hands and fingers. Subcutaneous granuloma annulare is characterized by large, skin-colored nodules that may be as deep as the lower dermis or subcutaneous fat.1
FIGURE 3.
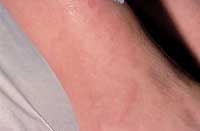
Granuloma annulare—plaques with raised nonscaly erythematous borders.
While the etiology of granuloma annulare is unknown,1,9 it has been hypothesized that it is caused by vasculitis, trauma, monocyte activation or type IV delayed hypersensitivity. Clear-cut evidence in support of one theory over another has yet to emerge.
Diagnosis is typically based on clinical appearance and correlation with pathology. Laboratory tests are generally of little benefit9; however, in patients with generalized granuloma annulare, abnormalities of glucose tolerance are more common.10
Because granuloma annulare is self-limited and ordinarily asymptomatic, the best treatment is no treatment.1 In patients concerned about cosmetic disfigurement and those who have symptomatic disease, many treatment possibilities exist, including: intralesional corticosteroid injections, topical corticosteroids, electrodesiccation, cryotherapy, ultraviolet light therapy and systemic agents, such as dapsone, colchicine or chloroquine (Aralen). Although these options have been shown to provide symptomatic improvement in some patients, none has a discernible advantage over the others, and there is currently no cure. It is difficult to gauge the effectiveness of any of these treatment options, considering the variability of the disease.
Sarcoidosis
Sarcoidosis is an idiopathic, multisystemic granulomatous disease first described by Hutchinson in 1877.11,12 Sarcoidosis is characterized by the formation of noncaseating granulomas in virtually every organ system in the human body, most commonly the lungs, skin, liver, spleen, eyes, salivary glands and lymph nodes, particularly the mediastinal nodes.12,13
The exact incidence of sarcoidosis is unknown.12 Although it can occur at any age, young adults between 20 to 40 years are at highest risk.11–13 In the United States, blacks have a higher incidence of sarcoidosis than whites and are more likely to have debilitating, widespread, chronic disease.11
The range of clinical presentation can vary from asymptomatic to fulminant disease. The typical skin changes may wax and wane and may include infiltrated papules and plaques, subcutaneous nodules and infiltration of old scars.12,13 The most common lesions are papular lesions on the face, especially in the periorbital area and the nasolabial folds,12 and even the mucous membranes.13 They typically appear as reddish-brown to purplish lesions that measure between 1 and 3 cm in diameter.12 There is generally little epidermal change.12 The lesions may be large enough to coalesce, which may give them the appearance of annular lesions or plaques (Figure 4).
FIGURE 4.
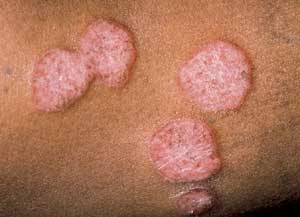
Sarcoidosis—indurated, erythematous plaques.
Diagnosis of sarcoidosis is based on clinical presentation, histology and radiology. Bilateral hilar lymphadenopathy is virtually specific to sarcoidosis.14 When sarcoidosis is suspected, a search for cutaneous involvement is vital because it allows for a biopsy and excludes the need for more aggressive diagnostic procedures. Bronchoalveolar lavage and transbronchial lung biopsy may also provide histologic evidence in 85 to 90 percent of patients.12,13
Although there is no cure for sarcoidosis, systemic corticosteroids effectively treat its symptoms.12,13 Indications for treatment include dyspnea, cough and widespread debilitating disease.12,14,15 Patients intolerant of or resistant to corticosteroids may benefit from therapy with hydroxychloroquine (Plaquenil), chloroquine, methotrexate (Rheumatrex) or thalidomide (Thalomid). Topical or intralesional corticosteroids may be beneficial for localized skin disease.13,15
Hansen's Disease
Hansen's disease, also known as leprosy, is rare in the United States outside of endemic areas such as Texas, Louisiana, Hawaii and California.16 The majority of cases in the United States occur in persons who have resided in foreign countries, particularly Asia, Africa and Latin America.16,17 The disease is caused by the acid-fast bacillus, Mycobacterium leprae, which is transmitted from person to person via droplets from the respiratory tract.16,17 Risk of transmission from casual and household contact is low.16 Although children are more vulnerable to infection than adults,16 95 percent of people are immune to Hansen's disease.18 The incubation period for this infection can run from three to 20 years.19
M. leprae grows best at 35.6°C (96.0°F) and thus has a predilection for cooler parts of the body, such as skin, peripheral nerves and eyes,18,19 with sparing of the groin, axillae and hairy scalp.16 Clinical presentation varies considerably depending on the host's response to infection.16,17 Patients with tuberculoid leprosy generally present with a few erythematous or violaceous, sharply demarcated macules or plaques (Figure 5), because of their healthy cellular immunity.16,17 These lesions are hypopigmented in persons with dark skin. The lesions may have associated scaling, alopecia and, most notably, anesthesia.19 Nerve damage occurs when the inflammatory cells attack the nerves that have been infiltrated by the bacteria, leading to loss of sensory and motor function, and a palpable nerve.16,19 Patients with lepromatous leprosy, however, suffer from defective cellular immunity17 and present with extensive, bilaterally symmetric macules and papules that may coalesce or become nodular. These patients have a high bacterial load.
FIGURE 5.
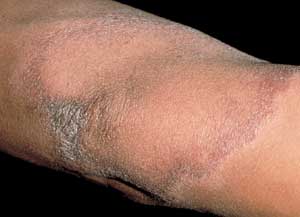
Hansen's disease—plaque with borders that can be associated with scales.
Clinical diagnosis depends on the history and pathology. Any hypoesthetic, hairless or dry lesion should immediately raise suspicion of Hansen's disease, particularly if there is an adjacent palpable nerve. Tuberculoid leprosy biopsies demonstrate epithelioid granulomas with abundant peripheral lymphocytes.17 Biopsies of lesions from patients with lepromatous leprosy will include many macrophages with foamy cytoplasm. When the diagnosis of Hansen's disease is suspected, the physician should request a special acid-fast stain (Fite's method) to identify the bacillus because the bacteria may lose its acid-fast characteristic when fixed and stained with Ziehl-Neelsen stain. While patients with tuberculoid leprosy have only a few organisms, those with lepromatous leprosy have abundant bacteria.16
A typical treatment course for Hansen's disease involves multiple drug therapy with dapsone and rifampin (Rifadin) for three to five years in patients with tuberculoid leprosy, and for life in patients with lepromatous leprosy.16
Urticaria
Urticaria is characterized by pruritic, well-circumscribed erythematous lesions of the skin (wheals) with erythematous raised borders and blanched centers (Figure 6). Urticaria may affect 10 to 20 percent of the population,20 but this figure may be an underestimation because the self-limited nature of this eruption may cause many patients to not seek medical attention. Following the release of histamine and other mediators, increased vascular permeability results in massive edema of the superficial dermis.
FIGURE 6.
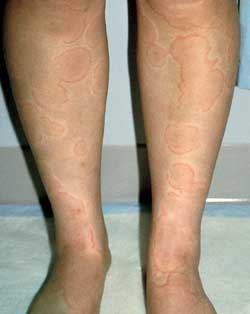
Urticaria—erythematous wheals with central clearing and no scale.
Based on the precipitating event, urticaria is classified as allergic, physical or idiopathic. Allergic urticaria may be caused by a different immunologic mechanism than the other types. IgE-mediated urticaria (type I, or immediate, hypersensitivity) results in degranulation of mast cells and histamine release. This mechanism is associated with urticaria caused by pollens, foods, medications, fungi, molds, Hymenoptera venom and parasitic infections. Antibody-dependent, cell-mediated cytotoxicity (type II hypersensitivity) and antigen-antibody complexes (type III hypersensitivity) and activation of the complement system account for the urticaria occurring in persons with transfusion reactions and serum sickness, respectively.
Physical urticaria includes pressure urticaria, which appears under sites of tight clothing, on the soles and wherever a heavy load is carried; cold urticaria, which usually occurs on the hands, ears, nose and the cooler fatty areas, such as lateral thighs in women; cholinergic urticaria, which is usually precipitated by fever, hot baths or exercise; solar urticaria; and dermograph urticaria. In most cases of urticaria, the specific etiology remains unclear.
A lesion of urticaria usually lasts between 90 minutes and 24 hours.21 Urticarial wheals can be greatly inhibited with the most potent antihistamines but usually cannot be totally suppressed, which suggests that histamine is not the only mediator.21 Urticarial lesions that persist for longer than 24 hours may represent urticarial vasculitis. Urticarial vasculitis is not actually urticaria but a leukocytoclastic vasculitis that can mimic urticaria clinically, with the exception that the lesions may persist up to three to five days. In these patients, a skin biopsy for histopathologic confirmation of vasculitis is warranted.
SCLE
Subacute cutaneous lupus erythematosus (SCLE) presents either in an annular (Figure 7) or a papulosquamous form. Photosensitivity is a major component of SCLE, and the lesions are generally confined to exposed surfaces. One half of these patients will fulfill four or more of the American Rheumatism Association's criteria for systemic lupus erythematosus (SLE). Patients with SLE commonly present with arthralgias or arthritis, low grade fever, malaise or myalgias. Systemic disease is mild to moderate, and the incidence of renal disease is low.
FIGURE 7.
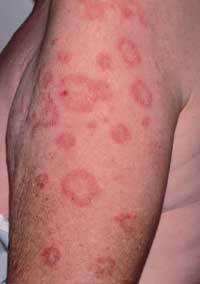
Subacute cutaneous lupus erythematosus. Annular erythematous plaques have central clearing, often mimicking annular psoriasis when associated with scales.
The Sjögren's syndrome antigen/Ro antibody (SSA/Ro) is present in 63 percent of patients with SCLE, and 5 to 10 percent of these patients have Sjögren's syndrome. It has been shown that SSA/Ro antibodies, which are present in the nucleus and cytoplasm, are translocated to the surface of the cultured keratinocytes under UVB light. Anti-SSA/Ro antibodies in the sera can then bind to the relevant antigens expressed on the UVB-light-irradiated keratinocyte surface and induce the skin lesions through a cytotoxic mechanism.22
Erythema Annulare Centrifugum
Erythema annulare centrifugum is characterized by non-indurated, annular patches with associated trailing scale inside the erythematous borders. Histologically, perivascular dermal lymphocytic infiltrates are gathered in a “coat-sleeve-like” appearance in the dermis, with concomitant papillary edema, spongiosis and parakeratosis.23,24 The annular lesions most commonly affect the trunk, buttocks, thighs and legs, while sparing the hands, feet and face.25 Erythema annulare centrifugum has a tendency to be recurrent, and there are rarely constitutional symptoms.23–25
The etiology and pathogenesis are unknown.23 It is believed that erythema annulare centrifugum represents a cutaneous manifestation of a hypersensitivity reaction to a myriad of underlying conditions, including: infection by dermatophytes, bacteria and viruses; malignancy; and immunologic disorders.1,23,25 However, available data suggest that there is no increased incidence of these associations with erythema annulare centrifugum in comparison to the general population.23,25
Diagnosis is clinicopathologic. The course of the disease is highly variable because erythema annulare centrifugum may last for as little as a few weeks or as long as three decades. Most cases persist for approximately nine months.23
Most cases do not require treatment. However, the underlying condition, if it is known, should be treated.1,25 Otherwise, physicians may give topical or systemic corticosteroids or antihistamines for symptomatic improvement of the pruritus.23,25
Other Annular Lesions
Erythema chronicum migrans is the cutaneous hallmark of Lyme disease. One or more large erythematous patches may appear anywhere on the skin. The lesions expand centrifugally, sometimes with central clearing, giving rise to annular patches.
Erythema multiforme is a cell-mediated immune reaction that is often related to herpes simplex infection. Cutaneous lesions characteristically appear on the palms, the soles, the elbows and the knees. Typically, a papule or plaque expands with erythematous borders, while the center may become necrotic or dusky, resulting in a “target lesion.”
Psoriasis, which most commonly presents as erythematous plaques with diffuse thick, white scale, can present as annular lesions with scale only on the borders (Figure 8).
FIGURE 8.
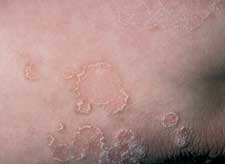
Psoriasis. Plaque-type psoriasis can present with scale only on the borders.
Nummular eczema is often associated with xerosis and commonly occurs on the legs. The lesions are coin-shaped papules and plaques with vesicles that may expand with central clearing, giving an annular appearance.
