Anemia in children is commonly encountered by the family physician. Multiple causes exist, but with a thorough history, a physical examination and limited laboratory evaluation a specific diagnosis can usually be established. The use of the mean corpuscular volume to classify the anemia as microcytic, normocytic or macrocytic is a standard diagnostic approach. The most common form of microcytic anemia is iron deficiency caused by reduced dietary intake. It is easily treatable with supplemental iron and early intervention may prevent later loss of cognitive function. Less common causes of microcytosis are thalassemia and lead poisoning. Normocytic anemia has many causes, making the diagnosis more difficult. The reticulocyte count will help narrow the differential diagnosis; however, additional testing may be necessary to rule out hemolysis, hemoglobinopathies, membrane defects and enzymopathies. Macrocytic anemia may be caused by a deficiency of folic acid and/or vitamin B12, hypothyroidism and liver disease. This form of anemia is uncommon in children.
Anemia is a frequent laboratory abnormality in children. As many as 20 percent of children in the United States and 80 percent of children in developing countries will be anemic at some point by the age of 18 years.1
Physiology of Hemoglobin Production
Erythropoietin is the primary hormone regulator of red blood cell (RBC) production. In the fetus, erythropoietin comes from the monocyte/macrophage system of the liver. Postnatally, erythropoietin is produced in the peritubular cells of the kidneys. Key steps in red blood cell differentiation include condensation of red cell nuclear material, production of hemoglobin until it amounts to 90 percent of the total red blood cell mass and the extrusion of the nucleus that causes loss of RBC synthetic ability. Normal RBCs survive an average of 120 days, while abnormal RBCs can survive as little as 15 days.1
The hemoglobin molecule is a hemeprotein complex of two pairs of similar polypeptide chains. There are six types of hemoglobin in developing humans: the embryonic, Gower-I, Gower-II, Portland, fetal hemoglobin (HbF) and normal adult hemoglobin (HbA and HbA2). HbF is the primary hemoglobin found in the fetus. It has a higher affinity for oxygen than adult hemoglobin, thus increasing the efficiency of oxygen transfer to the fetus. The relative quantities of HbF rapidly decrease to trace levels by the age of six to 12 months and are ultimately replaced by the adult forms, HbA and HbA2
General Approach to Management
Most children with anemia are asymptomatic and have an abnormal hemoglobin or hematocrit level on routine screening (Table 1).2 Infrequently, a child with anemia may have pallor, fatigue and jaundice but may or may not be critically ill. Key historical points and findings on physical examination can reveal the underlying cause of the anemia.
The newborn's body reclaims and stores iron as the hematocrit levels decrease during the first few months of life. Therefore, in full-term infants, iron deficiency is rarely the cause of anemia until after six months of age. In premature infants, iron deficiency can occur only after the birth weight has been doubled. X-linked causes of anemia, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency, should be considered in males. Pyruvate kinase deficiency is autosomal recessive and associated with chronic hemolytic anemia of variable severity. A history of nutritional deficiency, pica or geophagia suggests iron deficiency. Recent prescription drug use may suggest G6PD deficiency or aplastic anemia. A recent viral illness may suggest red cell aplasia. Recurrent diarrhea raises suspicion of malabsorption and occult blood loss occurring in celiac sprue and inflammatory bowel disease.
TABLE 1 Screening Recommendations for Anemia in Children
|
Information from U.S. Preventive Services Task Force. Guide to clinical preventive services: report of the U.S. Preventive Services Task Force. 2d ed. Baltimore, Md.: Williams & Wilkins, 1996; American Academy of Family Physicians. Summary of AAFP policy recommendations and age charts. Retrieved October 2000, from:https://www.aafp.org/exam; and the American Academy of Pediatrics. AAP policy statements, clinical practice guidelines, and model bills. Retrieved October 2000, from:http://www.aap.org/policy/pcyhome.cfm.
The physical examination is important but will be unremarkable in most children with anemia. Findings that suggest chronic anemia include irritability, pallor (usually not seen until hemoglobin levels are less than 7 g per dL [70 g per L]), glossitis, a systolic murmur, growth delay and nail bed changes. Children with acute anemia often present more dramatically with clinical findings including jaundice, tachypnea, tachycardia, splenomegaly, hematuria and congestive heart failure.
Laboratory Evaluation
Anemia is defined as a decreased concentration of hemoglobin and RBC mass compared with that in age-matched controls. In screening situations, such as the one-year check-up, only a hemoglobin level is usually obtained. When anemia is encountered during this screening, the specimen should be upgraded to a complete blood cell count (CBC), because some laboratories store blood samples for up to seven days. Physicians should first look at the mean corpuscular volume (MCV), which allows placement of the anemia into one of the standard classifications of microcytic, normocytic and macrocytic (Table 2).3,4 After narrowing the differential diagnosis based on the MCV, the clinician can proceed with additional diagnostic work-up.
The next step of the anemia work-up should include a peripheral smear and a measurement of the reticulocyte count. Pathologic findings on the peripheral smear can indicate the etiology of the anemia based on red cell morphology. Basophilic stippling (Figure 1a) representing aggregated ribosomes can be seen in thalassemia syndromes, iron deficiency and lead poisoning. Howell-Jolly bodies (Figure 1b) are nuclear remnants seen in asplenia, pernicious anemia and severe iron deficiency. Cabot's ring bodies (Figure 1c) are also nuclear remnants and are seen in lead toxicity, pernicious anemia and hemolytic anemias. Heinz's bodies (Figure 1d) are from denatured aggregated hemoglobin and can be seen in thalassemia, asplenia and chronic liver disease.
The reticulocyte count (or percentage) helps distinguish a hypoproductive anemia (decreased RBC production) from a destructive process (increased RBC destruction). A low reticulocyte count may indicate bone marrow disorders or aplastic crisis, while a high count generally indicates a hemolytic process or active blood loss. The corrected reticulocyte count corrects for differences in the hematocrit and is a more accurate indicator of erythropoietic activity. To calculate corrected reticulocyte count, multiply the patient's reticulocyte count (or percentage) by the result of dividing the patient's hematocrit level by the normal hematocrit level. A corrected reticulocyte count above 1.5 suggests increased RBC production. In the case of decreased RBC survival, the bone marrow normally responds with increased reticulocyte production, usually greater than 2 percent or with an absolute count of greater than 100,000 cells per mm3 (100 × 106 per L). This is presumptive evidence of chronic hemolysis if the reticulocytosis is sustained.
FIGURE 1.
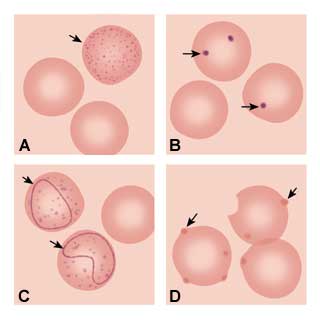
Depiction of red blood cell morphologies that may appear on a peripheral smear, showing: (A) basophilic stippling, (B) Howell-Jolly bodies, (C) Cabot's ring bodies and (D) Heinz's bodies.
If, after analysis of the initial laboratory findings, the diagnosis is still unclear, other confirmatory studies may be required. Tests to determine if the MCV is too low include serum iron level, total iron binding capacity (TIBC) and lead level. A serum ferritin level would be an acceptable substitute for the serum iron or TIBC levels. Serum ferritin levels are the first to decrease in patients with iron deficiency and are sensitive and specific. However, because serum ferritin is an acute phase reactant, it can be falsely elevated. If hemolysis is suspected, a direct Coombs' test, G6PD assay, hemoglobin electrophoresis, and lactate dehydrogenase (LDH), haptoglobin and bilirubin (indirect) determinations may help to confirm the diagnosis. For the anemic child with an elevated MCV, the physician should test the vitamin B12, folate and thyroid-stimulating hormone levels.
Other tests for diagnostic confirmation include an RBC enzyme panel to diagnose enzymopathies, osmotic fragility to diagnose hereditary spherocytosis, hemoglobin isoelectric focusing to diagnose hemoglobin variants, membrane protein studies to diagnose membranopathies, and cytogenetic studies.3 In certain circumstances, such as a suspected hematologic malignancy, a bone marrow aspiration may be indicated. Hematology consultation before ordering these more sophisticated tests is usually warranted.
Types of Anemia Based on the MCV
MICROCYTIC ANEMIAS
The most prevalent and preventable form of microcytic anemia is iron deficiency anemia.1 The prevalence of iron deficiency anemia in the United States ranges from 3 to 10 percent and may be as high as 30 percent in low-income populations.5 Researchers in a 1997 study6 of a private pediatric office in New York City evaluated 504 consecutive children, ages one to three years, for anemia. Children with a chronic or acute illness, premature birth or with a known blood dyscrasia were excluded from participating in the study. The authors found that approximately 7 percent of the children in this population were iron deficient without anemia and 10 percent had iron deficiency anemia.6
TABLE 3 Age-Specific Blood Cell Indexes
| Age | Hemoglobin, g/dL (g/L) | Hematocrit (%) | MCV, µm3 (fL) | MCHC, g/dL (g/L) | Reticulocytes | |
|---|---|---|---|---|---|---|
| 26 to 30 weeks' gestation* | 13.4 (134) | 41.5 (0.42) | 118.2 (118.2) | 37.9 (379) | — | |
| 28 weeks' gestation | 14.5 (145) | 45 (0.45) | 120 (120) | 31.0 (310) | (5 to 10) | |
| 32 weeks' gestation | 15.0 (150) | 47 (0.47) | 118 (118) | 32.0 (320) | (3 to 10) | |
| Term† (cord) | 16.5 (165) | 51 (0.51) | 108 (108) | 33.0 (330) | (3 to 7) | |
| 1 to 3 days | 18.5 (185) | 56 (0.56) | 108 (108) | 33.0 (330) | (1.8 to 4.6) | |
| 2 weeks | 16.6 (166) | 53 (0.53) | 105 (105) | 31.4 (314) | ||
| 1 month | 13.9 (139) | 44 (0.44) | 101 (101) | 31.8 (318) | (0.1 to 1.7) | |
| 2 months | 11.2 (112) | 35 (0.35) | 95 (95) | 31.8 (318) | ||
| 6 months | 12.6 (126) | 36 (0.36) | 76 (76) | 35.0 (350) | (0.7 to 2.3) | |
| 6 months to 2 years | 12.0 (120) | 36 (0.36) | 78 (78) | 33.0 (330) | ||
| 2 to 6 years | 12.5 (125) | 37 (0.37) | 81 (81) | 34.0 (340) | (0.5 to 1.0) | |
| 6 to 12 years | 13.5 (135) | 40 (0.40) | 86 (86) | 34.0 (340) | (0.5 to 1.0) | |
| 12 to 18 years | ||||||
| Male | 14.5 (145) | 43 (0.43) | 88 (88) | 34.0 (340) | (0.5 to 1.0) | |
| Female | 14.0 (140) | 41 (0.41) | 90 (90) | 34.0 (340) | (0.5 to 1.0) | |
| Adult | ||||||
| Male | 15.5 (155) | 47 (0.47) | 90 (90) | 34.0 (340) | (0.8 to 2.5) | |
| Female | 14.0 (140) | 41 (0.41) | 90 (90) | 34.0 (340) | (0.8 to 4.1) | |
MCV = mean corpuscular volume; MCHC = mean corpuscular hemoglobin concentration.
*— Values are from fetal samplings.
†— Less than one month, capillary hemoglobin exceeds venous: 1 hour–3.6 g difference; 5 days–2.2 g difference; 3 weeks–1.1 g difference.
Adapted with permission from Siberry GK, Iannone R, eds. The Harriet Lane handbook: a manual for pediatric house officers. 15th ed. St. Louis: Mosby, 2000.
Severe iron deficiency is usually easily diagnosable; however, the milder forms of iron deficiency offer a greater challenge. The normal values for the age-matched red cell indexes are listed in Table 37.
If the history and laboratory findings suggest iron deficiency anemia, a one-month empiric trial of iron supplementation is appropriate in asymptomatic infants nine to 12 months of age. A low MCV and elevated red cell distribution width (RDW) suggest iron deficiency.8 The RDW is an index of the variability in the size of the red blood cells (anisocytosis), which is the earliest manifestation of iron deficiency.9 Table 48 illustrates how the RDW helps distinguish iron deficiency from other causes of microcytosis.10
Iron supplements are given to the child at a dosage of 3 to 6 mg per kg per day in the form of ferrous sulfate before breakfast. An increase in hemoglobin levels of greater than 1.0 g per dL (10.0 g per L) by four weeks is diagnostic of iron deficiency anemia and warrants continuation of therapy for two to three additional months to properly replenish iron stores.11 During this time, further dietary intervention and patient education can be provided. If the anemia recurs, a work-up to identify the source of occult blood loss is warranted.
It is widely accepted that iron deficiency can have long-term consequences that are often irreversible. Several studies have found that reversal of the anemia did not improve standardized test scores.12,13 One study14 examined a group of Costa Rican children at five years of age. Children who had moderately severe iron deficiency anemia (hemoglobin less than 10 g per dL [100 g per L]) in infancy scored significantly lower on standardized tests at five years of age, despite a return to normal hematologic status and growth. Studies in rat models demonstrated that iron deficiency anemia in early life causes a deficiency in dopamine receptors that could not be corrected by reversing the anemia.15,16 It is therefore imperative that physicians attempt to prevent iron deficiency in children before the second year of life. Strategies for the prevention of iron deficiency anemia can reduce the chances of developing the disease (Table 517).
The indications listed in Table 48 can help differentiate the other microcytic anemias. Thalassemias are genetic deficiencies in the gene coding for globin chains. In patients with thalassemia, either the α-chain or the β-chain cannot be synthesized in sufficient quantities, lending to the nomenclature α-thalassemia or β-thalassemia. This deficiency produces an unbalanced globin chain synthesis that leads to premature RBC death (Table 618(p1403)). There are about 100 mutations of varying severity that cause thalassemia. They are more prevalent in persons of Mediterranean, African, Indian and Middle-Eastern descent. They cause disruption of hemoglobin polypeptide synthesis that can be asymptomatic, mildly symptomatic or cause severe anemia.
Referral is appropriate for cases in which the diagnosis is unclear and for treatment of the more severe types of anemia.
The clinician is often confronted with microcytic anemia in a population with a higher prevalence of thalassemias. The Mentzer index was developed to help distinguish thalassemia from iron deficiency. It is calculated by dividing the RBC count into the MCV. When the quotient is less than 13, thalassemia is more likely, and if the quotient is greater than 13, iron deficiency is more likely.19 Therefore, in the child with risk factors for iron deficiency, and a Mentzer index indicating iron deficiency, a trial of iron supplementation is warranted as outlined above. When the CBC is rechecked at four to six weeks, extra tubes of blood can be drawn and held depending on the CBC results. They can then be sent for hemoglobin electrophoresis or other clinically pertinent tests, if there has been an inadequate response to the iron supplementation trial.
TABLE 4 Relation of Red Cell Distribution Width and Mean Corpuscular Volume
| Red cell distribution width | Mean corpuscular volume | ||||
|---|---|---|---|---|---|
| Low | Normal | High | |||
| Normal (11.5 to 14.5)* | Heterozygous α- or ß-thalassemia | — | Aplastic anemia | ||
| Chronic disease | Normal | Preleukemia | |||
| High (greater than 14.5) | Iron deficiency, HgH disease or sickle-ß-thalassemia | Chronic disease | Folate deficiency | ||
| Red cell fragmentation | Liver disease | Vitamin B12 deficiency | |||
HgH = hemoglobin H.
*— Some Coulter counters may have varying normal ranges.
Reprinted with permission from Oski FA. Iron deficiency in infancy and childhood. N Engl J Med 1993;329:190–3.
Other causes of microcytic anemia are lead poisoning and sideroblastic anemia. Lead poisoning is diagnosed in a child with elevated serum lead level. The acquired and hereditary forms of sideroblastic anemia are very rare in children.
TABLE 6 Clinical and Hematologic Features of the Principal Forms of Thalassemia
| Type of thalassemia | Globin-gene expression | Hematologic features | Clinical expressions | Hemoglobin findings | |
|---|---|---|---|---|---|
| β-Thalassemias | |||||
| β° homozygous | β°/β ° | Severe anemia; normoblastemia | Cooley anemia | HbF greater than 90 percent No HbA HbA2 increased | |
| β+ homozygous | β+/β+ | Anisocytosis, poikilocytosis; moderately severe anemia | Thalassemia intermedia | HbA: 20 to 40 percent HbF: 60 to 80 percent | |
| β° heterozygous | β/β° | Microcytosis, hypochromia, mild to moderate anemia | May have splenomegaly, jaundice | Increases HbA2 and HbF | |
| β+ heterozygous | β/β+ | Microcytosis, hypochromia, mild anemia | Normal | Increased HbA2 and HbF | |
| β silent carrier, heterozygous | β/β+ | Normal | Normal | Normal | |
| δβ heterozygous | δβ/(δβ)° | Microcytosis, hypochromia, mild anemia | Usually normal | HbF: 5 to 20 percent HbA2: normal or low | |
| γδβ heterozygous | γδβ/(γδβ)° | Newborn: microcytosis hemolytic anemia normoblastemia Adult: similar to heterozygous δβ | Newborn: hemolytic disease with splenomegaly Adult: similar to heterozygous δβ | Normal | |
| α -Thalassemias | |||||
| α silent carrier | −, α/α,α | Mild microcytosis or normal | Normal | Normal | |
| α trait | −, α/−, α or −, − / α α | Microcytosis, hypochromia, mild anemia | Usually normal | Newborn: Hb Barts (γ4) 5 to 10 percent Child or adult: normal | |
| HbH disease | −, α /−, − | Microcytosis, inclusion bodies by supravital staining; moderately severe anemia | Thalassemia intermedia | Newborn: Hb Barts (γ4) 20 to 30 percent Child or adult: HbH (β4) 4 to 20 percent | |
| α−hydrops fetalis | −, −/ −, − | Anisocytosis, poikilocytosis; severe anemia | Hydrops fetalis; usually stillborn or neonatal death | Hb Barts (y4) 80 to 90 percent; no HbA or HbF | |
β= gene completely suppresses globin chain synthesis; β+ = gene produces a demonstrable globin chain product; HbF = fetal hemoglobin; HbA = normal adult hemoglobin; HbA2 = minor fraction of normal adult hemoglobin; HbH = hemoglobin H.
Adapted with permission from Nelson WE, Behrman RE, Kliegman R, Arvin AM, eds. Nelson Textbook of pediatrics. 15th ed. Philadelphia: Saunders,1996:1403.
NORMOCYTIC ANEMIAS
Determining a diagnosis of normocytic anemia in a child can be clinically difficult. First, obtain a reticulocyte count to determine whether there is decreased production or increased destruction of red blood cells. When there is increased destruction, the reticulocyte count will be high, the LDH and indirect bilirubin levels will increase, and there may be signs of red cell destruction on the peripheral smear (i.e., schistocytes, sickle cells, tear forms and poikilocytes). With decreased red cell production, the reticulocyte count will be depressed relative to the hemoglobin concentration. Depending on the severity of the anemia, the evaluation may ultimately warrant a bone marrow aspiration (Table 7).18(p1399)
The physiologic anemia of infancy is often confused with a pathologic condition. During the first weeks of life, erythropoietin synthesis abruptly decreases. In the ensuing six to eight weeks, the hemoglobin reaches a low point of 9 to 11 g per dL (90 to 110 g per L) or 7 to 9 g per dL (70 to 90 g per L) in premature infants, the erythropoietin production is again stimulated and the hemoglobin level is returned to normal. This often causes concern during the routine work-up of the febrile infant. A CBC obtained to evaluate the white blood cell count may reveal an “abnormal” hemoglobin level. This physiologic anemia, unless lower than the expected range for this age group, deserves no further work-up.
TABLE 7 Clinically Important Sickle Cell Syndromes
| Sickle cell disorder | Hemoglobin composition (%) | HbA2 level | Erythrocyte volume (MCV) | Clinical severity | Clinical features |
|---|---|---|---|---|---|
| HbSS | HbS: 80 to 95 | Normal | Normal | + + to + + + + | Severe disease |
| HbF: 2 to 20 | |||||
| HbS-β°-thalassemia | HbS: 75 to 90 | Increased | Decreased | + + to + + + + | Generally indistinguishable from SS |
| HbF: 5 to 25 | |||||
| HbS-β+-thalassemia | HbS: 5 to 85 | Increased | Decreased | + to + + + | Generally milder than SS |
| HbA: 10 to 30 | |||||
| HbF: 5 to 10 | |||||
| HbSS with α-thalassemia trait (−, α/ −, α) | HbS: 80 to 90 | Normal | Decreased | + + to + + + + | May be milder than SS |
| HbF: 10 to 20 | |||||
| HbSC | HbS: 45 to 50 | Normal | Normal | + to + + + | Generally milder than SS; higher frequency of bone infarcts and proliferative retinal disease |
| HbC: 45 to 50 | |||||
| HbF: 2 to 5 | |||||
| HbSo Arab | HbS: 50 to 55 | Normal | Normal | + + to + + + + | Generally indistinguishable from SS |
| HbO: 40 to 45 | |||||
| HbF: 2 to 15 | |||||
| HbSD Los Angeles | HbS: 45 to 50 | Normal | Normal | + + to + + + + | May be as severe as SS |
| HbD: 30 to 40 | |||||
| HbF: 5 to 20 | |||||
| HbS/HPFH* | HbS: 65 to 80 | Normal | Normal | 0 to + | Usually asymptomatic |
| HbF: 15 to 30 | |||||
| HbAS* | HbS: 32 to 45 | Normal | Normal | 0 to + | Asymptomatic |
| HbA: 52 to 65 |
HbA2 = normal adult hemoglobin; MCV = mean corpuscular volume; Hb = hemoglobin; HPFH = hereditary persisitence of fetal hemoglobin.
*— These conditions do not ordinarily produce sickle cell disease.
Adapted with permission from Nelson WE, Behrman RE, Kliegman R, Arvin AM, eds. Nelson Textbook of pediatrics. 15th ed. Philadelphia: Saunders, 1996:1399.
Infection with human parvovirus B19 (fifth disease) is a common cause of bone marrow suppression, typically causing four to eight days of aplasia.20 In healthy children, there are rarely hematologic complications; however, in children with sickle-cell disease or hereditary spherocytosis or elliptocytosis, the consequences of this viral-induced red cell aplasia can be catastrophic. This is because the average lifespan of a spherocyte or elliptocyte is markedly decreased from an average of 120 days to as low as 10 to 30 days. The circulating blood volume is therefore significantly more dependent on bone marrow production. Children with acute parvovirus infection are typically admitted to the hospital for intravenous immune globulin and blood transfusions if the anemia is symptomatic or severe (hemoglobin less than 3.5 g per dL [35 g per L]).21
Enzyme deficiencies, such as G6PD and pyruvate kinase are characterized by bouts of hemolysis during some form of stress. Deficiency of G6PD is the most common enzymopathy and is present in 13 percent of black males, 2 percent of black females and in some children of Mediterranean and Southeast Asian descent.22 In the case of G6PD deficiency, an oxidative stress may initiate a hemolytic anemia that can be dramatic. It will be manifested clinically by jaundice as well as other signs and symptoms of low hemoglobin levels. A reduced G6PD level will confirm the diagnosis but may be normal in the face of acute hemolysis. If this occurs, the test should be repeated several months after resolution of the episode. Currently, most hospitals test for G6PD and pyruvate kinase as part of newborn screening before discharge from the hospital.
MACROCYTIC ANEMIAS
Macrocytic anemias in children are relatively uncommon, but are usually caused by a deficiency of vitamin B12 and folate. Other possible causes include chronic liver disease, hypothyroidism and myelodysplastic disorders.
Folic acid deficiency is usually a secondary cause to inadequate dietary intake. Human and cow's milk provide adequate sources of folic acid. The treatment of this deficiency is with parenteral or oral folate in a dosage of 1 to 3 mg once daily.23 A hematologic response to folate supplementation can be seen within 72 hours.
Vitamin B12 deficiency from nutritional deprivation is rare in the United States. Congenital pernicious anemia arises from the inability to secrete the gastric intrinsic factor. Neurologic symptoms become present at about nine months of age depending on vitamin B12 stores from birth.4 The preferred treatment is lifelong vitamin B12 supplementation.
Final Comment
The treatment modalities and diagnostic work-up for anemias in children have been well delineated. One major area for improvement in primary care is the prevention of iron deficiency, because it has been associated with permanent delays in psychomotor development. Appropriate screening and subsequent diagnostic testing will allow the family physician to appropriately diagnose most cases of anemia in children. Hematology referral is always appropriate for complicated or less defined cases.
