Dermatomyositis is an idiopathic inflammatory myopathy with characteristic skin manifestations. Although the disorder is rare, with a prevalence of one to 10 cases per million in adults and one to 3.2 cases per million in children, early recognition and treatment are important ways to decrease the morbidity of systemic complications. An association with other connective tissue disorders (overlap syndrome) and malignancy make this diagnosis particularly important to primary care physicians. Patient management includes careful evaluation for underlying malignancy and liberal use of physical therapy, antihistamines, sunscreen and oral corticosteroids. Poor prognostic indicators include poorly responsive disease, delay in diagnosis and the presence of malignancy. The therapeutic goal is to maintain function and prevent or minimize sequelae.
Dermatomyositis is an idiopathic disorder that includes an inflammatory myopathy and characteristic skin manifestations; polymyositis includes the inflammatory myopathy without the cutaneous findings. The etiology of dermatomyositis remains unknown; some studies have reported an association with histocompatability antigens, environmental agents (e.g., virus, drugs) and autoimmunity.1 The average age at diagnosis is 40, and almost twice as many women are affected as men.2 The average age of onset in juvenile dermatomyositis is between five and 14 years. This subgroup of patients has a better prognosis than adult patients. Modern therapy has reduced mortality from near 50 percent to less than 10 percent.3
TABLE 1 Classification of Dermatomyositis/Polymyositis
| Dermatomyositis | ||
| Without muscle weakness (amyopathic dermatomyositis or dermatomyositis sine myositis) | ||
| With muscle weakness | ||
| Adult | ||
| Associated with cancer | ||
| Not associated with cancer | ||
| Pediatric | ||
| Polymyositis | ||
| Adult | ||
| Pediatric | ||
| Inclusion-body myositis | ||
| Overlap (myositis associated with a connective tissue disease) | ||
Adapted with permission from Drake LA, Dinehart SM, Farmer ER, Goltz RW, Graham GF, Hordinsky MK, et al. Guidelines of care of dermatomyositis. J Am Acad Dermatol 1996;34(5 pt 1):824–9.
Diagnostic Criteria
Classification of dermatomyositis and polymyositis was first described in 19754,5 and has been only slightly revised to include amyopathic dermatomyositis6–8 (Table 1).2 Based on the classification system, diagnostic criteria were developed to further assist in the confidence of the diagnosis (Table 2).9 The differential diagnosis of dermatomyositis is listed in Table 3.
Manifestations
CUTANEOUS
Cutaneous manifestations of dermatomyositis are generally grouped as pathognomonic, characteristic, compatible, less common and rare (Table 4). The primary lesion appears as a violaceous, macular erythema with a symmetric distribution. This may progress and become poikilodermatous (atrophic with telangiectasia and pigmentary changes) and indurated (as a result of mucin deposition).3 Pathognomonic manifestations include Gottron's papules and Gottron's sign (Figure 1). Gottron's papules, violaceous papules overlying the dorsal interphalangeal or metacarpophalangeal areas, elbow or knee joints, occur in approximately 70 percent of patients with dermatomyositis.3 Gottron's sign is erythematous or violaceous, often atrophic, macules or plaques in the same symmetric distribution pattern but sparing the interphalangeal spaces—just the opposite dermatologic distribution pattern on the hand that is observed in patients with systemic lupus erythematosus.
TABLE 2 Classification Criteria for Polymyositis and Dermatomyositis*
| 1. Skin lesions | |
| Heliotrope: red-purple edematous erythema on the upper palpebra | |
| Gottron's sign: red-purple keratotic, atrophic erythema or macules on the extensor surface of finger joints | |
| Erythema on the extensor surface of extremity joints, slight raised red-purple erythema over elbows or knees | |
| 2. Proximal muscle weakness (upper or lower extremity and trunk) | |
| 3. Elevated serum creatine kinase or aldolase level | |
| 4. Muscle pain on grasping or spontaneous pain | |
| 5. Myogenic changes on electromyography (short-duration, polyphasic motor unit potentials with spontaneous fibrillation potentials) | |
| 6. Positive anti-Jo-1 antibody test (histidyl-tRNA synthetase) | |
| 7. Nondestructive arthritis or arthralgias | |
| 8. Systemic inflammatory signs (temperature: more than 37°C [98.6°F] at axilla, elevated serum C-reactive protein level or accelerated erythrocyte sedimentation rate of more than 20 mm per hour by Westergren) | |
| 9. Pathologic findings compatible with inflammatory myositis (inflammatory infiltration of skeletal evidence of active regeneration may be seen) | |
*— Patients presenting with at least one finding from item 1 and four findings from items 2 through 9 are said to have dermatomyositis (sensitivity, 94.1 percent [127/135] and specificity of skin lesions against systemic lupus erythema and systemic sclerosis, 90.3 percent [214/237]). Patients presenting with at least four findings from items 2 through 9 are said to have polymyositis (sensitivity, 98.9 percent [180/182] and specificity of polymyositis and dermatomyositis against all control diseases combined, 95.2 percent [373/392]).
Adapted with permission from Tanimoto K, Nakano K, Kano S, Mori S, Ueki H, Nishitani H, et al. Classification criteria for polymyositis and dermatomyositis. J Rheumatol 1995;22:4.
Heliotrope, a macular rash with periorbital edema, is considered a characteristic finding of dermatomyositis, as are periungual telangiectasias (Figure 2).1,8 The rash occurs early in the course of the disease in 30 to 60 percent of patients.3 The characteristic lesions of the shawl sign and the V-sign appear as erythematous, poikilodermatous macules distributed in a “shawl” pattern over the shoulders, arms and upper back (Figure 3) and in a V-shaped distribution over the anterior neck and chest.10 Mechanic's hand (Figure 4) and may be associated with an increased risk of interstitial lung disease.10–12
TABLE 3 Differential Diagnosis of Dermatomyositis
| HIV infection (at onset of immunodeficiency) |
| Lichen planus |
| Polymorphous light eruption |
| Seborrheic dermatitis |
| Systemic lupus erythematosus |
| Psoriasis |
| Contact dermatitis |
| Atopic dermatitis |
| Trichinosis (caused by periorbital swelling and edema) |
| Alcohol |
| Drug effects* |
| Penicillamine, nonsteroidal anti-inflammatory agents (nifluric acid and phenylbutazone), hydroxyurea (Hydrea), pravastatin (Pravachol), clofibrate (Atromid-S) and ipecac |
HIV = human immunodeficiency virus.
*— A partial listing of drugs that can cause myositis.
Poikiloderma atrophicans vasculare (poikilodermatomyositis), a circumscribed violaceous erythema with associated telangiectasia, hypopigmentation and superficial atrophy, is most commonly found over the posterior shoulders, back, buttocks and a V-shaped area of the anterior neck and chest, and is often a late finding.10 Calcium deposition (calcinosis cutis) occurs in approximately 30 to 70 percent of cases of juvenile dermatomyositis and in only 10 percent of adult cases.13–15 The calcinosis is most commonly present on the buttocks, elbows, knees or traumatized areas, and is associated with increased disease activity and duration. Skin findings may be subtle even in patients with severe myositis and are not a measure of the severity of the disease.
TABLE 4 Cutaneous Manifestations of Dermatomyositis
| Pathognomonic manifestations |
| Gottron's papules: violaceous Erythematous papules overlying the dorsal interphalangeal or metacarpophalangeal, elbow or knee joints |
| Gottron's sign: symmetric, nonscaling, violaceous Erythematous macules or plaques, often atrophic, in the same distribution as Gottron's papules |
| Characteristic manifestations |
| Shawl sign/V-sign |
| Heliotrope |
| Periungual telangiectasias |
| Mechanic's hand |
| Compatible manifestations |
| Poikiloderma atrophicans vasculare |
| Calcinosis cutis |
| Less common Manifestations |
| Facial swelling |
| Malignancy |
| Erythroderma |
| Lichen planus |
| Cutaneous vasculitis |
| Panniculitis |
| Rare manifestations |
| Follicular hyperkeratosis |
| Papular mucinosis |
| Hypertrichosis |
| Malignant erythema |
| Urticaria/urticarial vasculitis |
| Partial lipodystrophy |
| Malignant atrophic papulosis (Degos' disease) |
| Zebra-like striping |
| Vulvar/scrotal involvement |
FIGURE 1.
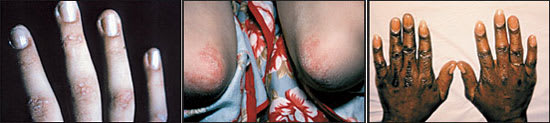
Pathognomonic manifestations of dermatomyositis. (Top) Gottron's papules overlying the dorsal interphalangeal joints. (Middle) Gottron's papules on the elbow. (Bottom) Gottron's sign: erythematous or violaceous atrophic macules and plaques overlying the dorsal interphalangeal joints and sparing the interphalangeal spaces.
FIGURE 2.
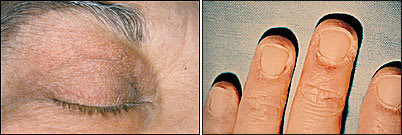
Characteristic findings of dermatomyositis. (Top) Heliotrope: a violaceous eruption with periorbital edema. (Bottom) Periungual telangiectasias.
FIGURE 3.
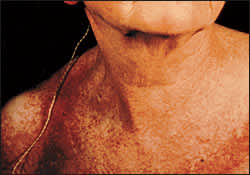
Shawl sign. Poikilodermatous macules appear in a “shawl” distribution over the shoulder, arms and upper back.
FIGURE 4.
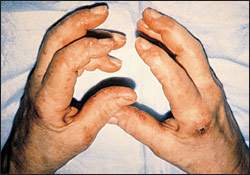
Mechanic's hand. Fissured, scaly, hyperkeratotic and hyperpigmented hands are suggestive of manual labor.
SYSTEMIC
Patients with dermatomyositis may also present with many systemic symptoms (Table 5). The most common are proximal muscle weakness, dysphonia or dysphagia. Other possible symptoms include respiratory muscle weakness, visual changes and abdominal pain. An important association with internal malignancy has been demonstrated and will be discussed in further detail.
TABLE 5 Systemic Manifestations and Complications of Dermatomyositis
| Systemic manifestations |
| Common: proximal muscle weakness, dysphonia, dysphagia |
| Less common: respiratory muscle weakness, visual changes, abdominal pain |
| Systemic complications/associations |
| Cardiomyopathy |
| Cardiac conduction defects |
| Aspiration pneumonia secondary to respiratory muscle weakness |
| Diffuse interstitial pneumonitis/fibrosis |
| Large-bowel infarction secondary to vasculopathy has occurred in juvenile patients with myositis |
| Muscle atrophy |
| Muscle calcification |
| Ocular complications including iritis, nystagmus, cotton-wool spots, optic atrophy, conjunctival edema and pseudopolyposis |
| Internal malignancy |
Subtypes of Dermatomyositis
JUVENILE DERMATOMYOSITIS
While the clinical presentation of juvenile dermatomyositis is usually different from the presentation of the adult type, the skin lesions are similar, with the exception of an increased incidence of calcinosis cutis in juvenile patients. Common findings include low-grade fever, increased risk of gastrointestinal manifestations, and symmetric arthritis of the large and small joints.16,17 Asymptomatic cardiac conduction delays or right bundle branch block may be found in 50 percent of this group.18
Patients may exhibit weakness of the truncal muscles that requires them to use their arms to push themselves up from a prone position (i.e., Gower's sign). There does not appear to be any association between juvenile dermatomyositis and malignancy.3
OVERLAP SYNDROME
A number of patients with dermatomyositis also meet the criteria for one of the connective tissue disorders. To be a true overlap syndrome, the patient must meet the diagnostic criteria for each separate disorder. Overlap syndrome occurs more frequently in females than in males, with a 9:1 ratio.3 Eleven to 40 percent of patients with dermatomyositis have been reported to have a concomitant diagnosis of a connective tissue disorder.3,19–21
These disorders include rheumatoid arthritis, scleroderma, systemic lupus erythematosus, Sjögren's syndrome, polyarteritis nodosa and mixed connective tissue disease, and these patients may present with polyarthritis, sicca syndrome, sclerodactyly, Raynaud's phenomenon and late symptoms of myositis. Patients are also more likely to have positive nonmyositis–associated antibodies (such as double-stranded DNA, antinuclear antibodies [ANA], Scl-70, Jo-1 precipitating antibodies, PM-Scl, Ku antibodies or extractable nuclear antigen antibodies). ANA are found in up to 80 percent of patients with dermatomyositis or polymyositis, but this finding does not aid in distinguishing myositis from scleroderma or other rheumatologic diseases.3,10 Precipitating autoantibodies to the Mi-2 antigen are specific for dermatomyositis but are found in only about 20 percent of patients with dermato-myositis.2 In patients with overlap syndrome, the myositis tends to respond better to treatment with corticosteroids than it does in patients with an idiopathic etiology.22
AMYOPATHIC DERMATOMYOSITIS
This classification has been controversial because it does not strictly meet the criteria put forth by Bohen and Peter.4,5 Amyopathic patients essentially have pathognomonic skin changes without clinical or laboratory evidence of muscle involvement. This condition has been reported in approximately 2 to 11 percent of patients with dermatomyositis.7,21,23 Patients most commonly present with lethargy, pruritus, fatigue, photosensitivity or arthralgias.8 In some cases, myositis developed later; in others, myositis that was not found by standard methods was suspected on the basis of magnetic resonance imaging (MRI).
DERMATOMYOSITIS/MALIGNANCY
Although an increased risk of malignancy has not been associated with juvenile dermatomyositis, it has been demonstrated in adults with dermatomyositis. One study suggested a 6.5-fold increased risk of malignancy.24 This risk appears to be highest in patients diagnosed with dermatomyositis after 45 years of age. The most commonly reported malignancies are ovarian and gastric cancer, and lymphoma. Other reported malignancies include lung, male genital organ, nonmelanoma skin, Kaposi's sarcoma, mycosis fungoides and melanoma.25,26
Skin changes are not different in patients with or without malignancy. Therefore, careful investigation for malignancy should be initiated at the time dermatomyositis is diagnosed. In women with dermatomyositis, there is a significant association with ovarian cancer, and some authors recommend that the work-up for dermatomyositis include a comprehensive gynecologic evaluation, including a cancer antigen (CA-125) baseline screen, mammography and transvaginal ultrasonographic evaluation of the ovaries at baseline, and gynecologic examinations at six- to 12-month intervals for at least two years.2,27
Evaluation
A complete history should be obtained and a physical examination performed, including a thorough review of systems, with an emphasis on myositis-related presentations and evidence of skin changes. Recommended tests that should be performed during the initial evaluation are listed in Table 6.2 Common laboratory manifestations of dermatomyositis are listed in Table 7.
TABLE 6 Evaluation of Dermatomyositis
| History and physical examination |
| Chest radiograph |
| Serum aldolase, LDH, ALT, AST, CBC with differential, CK, ANA/ENA in appropriate clinical setting, routine serum chemistry |
| Stool guaiac (three samples) |
| Urinalysis, urine myoglobin |
| Muscle biopsy |
| Skin biopsy* |
| Electromyography |
LDH = lactic dehydrogenase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CBC = complete blood count; CK = creatine kinase; ANA = antinuclear antibody; ENA = extractable nuclear antigens.
*— Skin biopsy findings of dermatomyositis are not specific for dermatomyositis but may help exclude other skin conditions that can have clinical appearances similar to the early cutaneous changes of dermatomyositis. 2
Treatment
The goal is to improve function and prevent disability. The treatment regimen must be instituted early and requires a team approach between the physical therapist, dermatologist and family physician. Other subspecialist involvement may be required, depending on the particular manifestations of the disease. Before the development of treatment, mortality from complications of dermatomyositis was approximately 50 percent, so patient education is particularly important.3
NONPHARMACOLOGIC AND TOPICAL THERAPY
Physical therapy is directed at preventing atrophy and contractures, and is particularly necessary in patients with calcinosis and muscle involvement. Technique should focus initially on passive stretching and splinting, with inclusion of more aggressive strength-building therapy once inflammation is controlled. The use of a broad-spectrum sunscreen is recommended in all patients with dermatomyositis and has the greatest benefit in patients who are photosensitive. Sun-avoidance techniques should be used, including the use of protective clothing. For control of severe pruritus, antihistamines (such as hydroxyzine [Atarax] or doxepin [Sinequan]) are recommended. For further control of the erythematous and pruritic skin changes, a class I (super-high potency) or class II (high potency) topical corticosteroid is recommended.3,10
TABLE 7 Laboratory Manifestations of Dermatomyositis
| Muscle enzyme elevation | |
| Serum aldolase, ALT, LDH, CK,* AST, carbonic anhydrase isoenzyme III† | |
| Autoantibodies | |
| ANA levels: elevated in 60 to 80 percent of patients with classic DM/PM | |
| Antisynthetase antibodies | |
| Jo-1: most common antisynthetase found; 20 percent of patients with DM may have positive result | |
| Anti-EJ: may be more associated with typical skin lesions | |
| SRP: occurring in 5 percent of patients, associated with polymyositis, acute-onset, severe, treatment-resistant forms of classic DM/PM with cardiac involvement | |
| Mi-2 antibodies (a nuclear protein complex): occurring in 15 to 20 percent of patients with classic DM, associated with a more treatment-responsive form, shawl sign and prominent cuticular changes | |
| Anti-PM-Scl antibodies: associated with overlap of scleroderma and DM/PM | |
| Anti-Ku antibodies: associated with overlap of scleroderma or SLE with DM/PM | |
| ESR: elevated in approximately 50 percent of patients, does not correlate well with disease activity | |
| Rheumatoid factor: elevated in 20 percent of patients, most often in those with overlap syndromes | |
| Neopterin and factor VIII–related antigen (von Willebrand factor): reported to correlate with juvenile DM | |
| EMG: myopathic pattern, 10 percent are false-negative | |
CK = creatinine kinase; AST = aspartate aminotransferase; ALT = alanine aminotransferase; LDH = lactic dehydrogenase; ANA = antinuclear antibody; DM = dermatomyositis; PM = polymyositis; SRP = signal recognition particle; SLE = systemic lupus erythematosus; ESR = erythrocyte sedimentation rate; EMG = electromyography.
*— Used to follow course of disease.
†— More sensitive but not widely available.
SYSTEMIC PHARMACOLOGIC THERAPY
Table 83,10 lists treatment modalities. Prednisone remains the initial oral pharmacologic agent, given in a single daily dose of 0.5 to 1.5 mg per kg until the serum creatine kinase (CK) level is normalized and then slowly tapered over the following 12 months. An alternate regimen is 40 to 60 mg of prednisone per day (1 to 2 mg per kg in children) in divided doses until the CK level has normalized, at which point the drug can be consolidated to a single daily dose. If the CK level remains within normal limits, that dose may be reduced by one fourth every three to four weeks. Prednisone should continue to be tapered until a maintenance dosage of 5 to 10 mg per day is reached. This low dosage should be continued for one year. If the CK level increases, a higher dosage of prednisone is warranted. If no improvement in objective muscle strength occurs after three months of prednisone therapy, other immunosuppressive therapy should be considered. The latter will be necessary in approximately 25 percent of patients.3,10
It is important to be aware of steroid myopathy, which may mimic a worsening dermatomyositis. Differentiating steroid myopathy from worsening dermatomyositis is based on evaluation of neck flexor strength—neck flexor strength would be unchanged if steroid myopathy were developing.3,10
Methotrexate (Rheumatrex) is considered the first-line adjuvant therapy in patients who do not respond to prednisone. Oral therapy should be initiated at a dosage of 7.5 to 10 mg per week and increased by 2.5-mg increments until a goal of 25 mg per week is reached. In children, a dosage of 1 mg per kg has been used. As the methotrexate dosage increases, the dosage of steroid should be decreased. Alternate intravenous dosing initiated at 10 mg per week should be increased by 2.5 mg per week until a total dosage of 0.5 to 0.8 mg per kg is reached. It is important to also give 1 to 3 mg per day of folic acid to minimize the side effects of methotrexate. Adverse effects include stomatitis, hepatic fibrosis, cirrhosis, nausea, abdominal pain, neutropenia, pruritus, fever, pneumonitis and other gastrointestinal symptoms. A liver biopsy should be considered before treatment is initiated. Methotrexate should be used only by physicians who are familiar with the drug's actions and side effects.3,10
Serum immunoglobulin has been used with success for treatment of patients with refractory dermatomyositis.28 Because of the limited duration of improvement and its high cost, this agent has not become a primary therapy.
Prognosis
Poor prognostic indicators include recalcitrant disease, delay in diagnosis, older age, malignancy, fever, asthenia-anorexia, pulmonary interstitial fibrosis, dysphagia and leukocytosis. Malignancy, cardiac and pulmonary dysfunction, and infection are the most common causes of death. With early treatment, survival rates as high as 80 and 73 percent at five and eight years, respectively, have been reported.29 Poor prognostic indicators in juvenile dermatomyositis are late onset of treatment, initial treatment with a dosage of prednisone that is too low, recalcitrant disease and pharyngeal involvement. Up to two thirds of this patient population develop severe complications of calcinosis cutis13,30 with mortality rates between 3 and 10 percent.10,18
TABLE 8 Treatment Modalities for Dermatomyositis
| Treatment modality | Dosage | Side effects | Comments |
|---|---|---|---|
| Oral prednisone | 0.5 to 1.5 mg per kg daily until serum creatine kinase normalizes, then slowly taper over 12 months | Gastrointestinal symptoms, adrenal suppression, immunosuppression, avascular necrosis, osteoporosis | Initial pharmacologic agent; consider adjunctive therapy if no improvement in objective muscle strength after three months of therapy. |
| Methotrexate (Rheumatrex) | Oral: 7.5 to 10 mg per week, increased by 2.5 mg per week to total of 25 mg per week Intravenous: 10 mg per week, increased by 2.5 mg per week to total of 0.5 to 0.8 mg per kg Children: 1 mg per kg. As dosage increases, taper off steroid dose. Give 3 mg daily of folic acid to minimize side effects of methotrexate. | Stomatitis, hepatic fibrosis, cirrhosis, nausea, abdominal pain, neutropenia, thrombocytopenia, pruritus, fever, pneumonitis, and gastrointestinal symptoms | First-line adjuvant therapy in patients unresponsive to steroids. Pretreatment liver biopsy for those with underlying liver disease. |
| Azathioprine (Imuran) | 2 to 3 mg per kg per day tapered to 1 mg per kg per day once steroid is tapered to 15 mg per day. Reduce dosage monthly by 25-mg intervals. Maintenance dosage is 50 mg per day. | Lymphoma, nausea, vomiting, hepatotoxicity, leukopenia, oral ulcers, thrombocytopenia | Screen patients for thiopurine methyltransferase deficiency before therapy (0.3 to 11 percent of white population). |
| Cyclophosphamide (Cytoxan) | Oral: 1 to 3 mg per kg per day Intravenous: 2 to 4 mg per kg per day, in conjunction with prednisone | Increased risk for malignancy, leukopenia, thrombocytopenia, hemorrhagic cystitis, anorexia, nausea, vomiting, alopecia, sterility, congestive heart failure and stomatitis | In refractory cases only |
| Cyclosporine (Sandimmune) | 2.5 to 10 mg per kg per day* | Impairs T-cell proliferation; nephrotoxicity, lymphoma, hypertension, hypertrichosis, gingival hyperplasia, hepatotoxicity, paresthesias, fatigue, hyperesthesias, depression and seizures | Maintain whole blood level of 200 to 300 ng per mL; may have rapid response to therapy |
| Hydroxychloroquine (Plaquenil) | 200 mg twice daily in adults; 2 to 5 mg per kg per day in children | Myopathy, differentiated by biopsy; hematologic toxicity, hepatotoxicity, antimalarial retinopathy, dizziness, ataxia and weight loss | Adjunctive treatment to reduce rash |
| Intravenous immunoglobulin | 2 g per kg in divided doses once per month for three months | Pancytopenia, death, lymphoma | Showed improvement in 70 percent of patients; limited by high cost |
| Total body irradiation | 15 rads biweekly over five weeks for total of 150 rads | Pancytopenia, death, lymphoma | Only case studies |
| Topical steroids | Class I (super-high potency) or class II (high potency) topical steroid is recommended | — | For further control of the erythematous and pruritic skin changes |
| Physical therapy | — | — | Directed at preventing atrophy andcontractures; technique should focus initially on passive stretching and splinting; more aggressive strength-building therapy once inflammation is controlled |
| Sun avoidance | — | — | Broad-spectrum sunscreen, protective clothing, avoiding ultraviolet light exposure |
| Antihistamines | — | — | For control of severe pruritus |
| Thymectomy and plasmapheresis | — | — | Investigational |
*— Cyclosporine dosing is highly subjective; it is used only as an adjunct to oral steroid therapy in a dosage of 2.5 to 10 mg per kg per day, then tapered to the lowest effective dosage over two weeks.
Information from Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol 1998;39:899–913, and Sontheimer RD. Dermatomyositis. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick's Dermatology in general medicine. 5th ed. New York: McGraw-Hill, 1999:2009–20.
