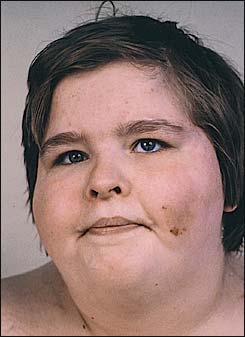
A 17-year-old mentally retarded, morbidly obese girl had a long history of sleep apnea, with excessive daytime sleepiness and loud snoring. On physical examination, notable findings included a height of 141 cm (56.4 in, well below the 5th percentile for age) and weight of 170 kg (374 lb, far greater than the 95th percentile for age). Abnormal facial features included strabismus, narrow nose, thin upper lip, down-turned mouth, viscous saliva, and dental caries (Figure 1). The patient habitually picked at her skin. Brilliant Green, an antiseptic dye, had been applied to multiple excoriations of the skin. Her skin and hair were lighter in color than those of other members of her family. Massive lymphedema of the lower extremities and small hands and feet were noted (Figure 2).
FIGURE 1.
Question
Based on the patient's history and physical examination, which one of the following is the most likely diagnosis?
A. Morbid obesity.
B. Pickwickian syndrome.
C. Prader-Willi syndrome.
D. Angelman syndrome.
Discussion
The answer is C: Prader-Willi syndrome. First described in 1956,1 it is the most common genetic cause of marked obesity, affecting one in 16,000 births.2 The clinical features of this disorder are often subtle and nonspecific, making the diagnosis difficult, particularly in infancy. Major diagnostic criteria include infantile hypotonia, feeding difficulties in infancy followed by excessive weight gain after 12 months of age, typical facial features, developmental delay, and hypogonadism.
Prader-Willi syndrome should be considered in the differential diagnosis of infantile hypotonia. The hypotonia is central, often severe, and is associated with a weak cry and suck. Poor feeding is a characteristic feature, which results in failure to thrive in the majority of infants with this syndrome. Deep tendon reflexes may be decreased or absent. The hypotonia tends to improve over time in weeks, months or, on occasion, one to three years after birth, although mild hypotonia persists throughout life.3 In contrast to the poor weight gain that is characteristic in early infancy, obesity usually becomes a problem between one and four years of age.
Typical facial features, more apparent after infancy, include a narrow face, almond-shaped eyes, thin upper lip with down-turned corners of the mouth, and a narrow nasal bridge. Strabismus is present in the majority of persons with Prader-Willi syndrome.
Hypogonadism may be difficult to assess before puberty in females, but in males, penile length and undescended testes or small testicular size may be found on examination. Young children have delayed motor and language development; mild to moderate mental retardation or learning problems are typical in older children.4 Minor criteria of Prader-Willi syndrome include sleep apnea, hypopigmentation of the skin and hair compared with other family members, small hands and feet, viscous saliva, and skin picking.4
Prader-Willi syndrome and Angelman syndrome are distinct disorders that are caused by the loss of the function of genes located on the long arm of chromosome 15. Angelman syndrome is often characterized by more severe mental retardation and only rarely by obesity. In Prader-Willi syndrome, the genetic loss occurs on the paternally inherited chromosome 15, while in Angelman syndrome, the loss is of maternal origin. DNA methylation analysis can distinguish between the maternally and paternally inherited chromosome 15, and is useful in screening for Prader-Willi and Angelman syndromes.5
Morbid obesity is present in this patient but does not explain the dysmorphic facial features, skin picking, and mental retardation that also are noted. Pickwickian syndrome describes the severe sleep apnea, hypercapnia, and daytime somnolence that may occur in very obese patients, but again would not by itself account for the other features seen here.
