To complement the 2005 Annual Clinical Focus on medical genomics, AFP will be publishing a series of short reviews on genetic syndromes. This series was designed to increase awareness of these diseases so that family physicians can recognize and diagnose children with these disorders and understand the kind of care they might require in the future. The first review in this series discusses fragile X syndrome.
Fragile X syndrome is caused by an expansion mutation in the Fragile X mental retardation 1 (FMR1) gene located on the X chromosome It characteristically leads to some degree of mental retardation. The phenotype is subtle, with minor dysmorphic features and developmental delay during childhood. Characteristic features during adolescence are an elongated face, prominent jaw, large ears, macro-orchidism, and a range of behavioral anomalies and cognitive deficits (Figure 1). Recently recognized manifestations in premutation carriers include premature ovarian failure and tremor/ataxia.1 Premature ovarian failure occurs in up to 20 percent of women who are premutation carriers of the FMR1 gene.1 Fragile X–associated tremor/ataxia syndrome (FXTAS) affects 30 percent of premutation carrier men between the ages of 50 and 60 years, and its prevalence increases with age.1
Figure 1.

Dysmorphic findings in persons with fragile X syndrome include an elongated face, prominent jaw, and large ears.
Epidemiology
Fragile X syndrome, the phenotype associated with full mutation, occurs in approximately one in 4,000 men and one in 6,000 to 8,000 women. The premutation in the FMR1 gene occurs in approximately one in 800 men and up to one in 100 to 200 women. Premutation male carriers are susceptible to FXTAS after age 50. Premature ovarian failure may be detected in as many as one third of women with a premutation.
Clinical Presentation
Although fragile X syndrome occurs in males and females, females generally present with milder symptoms. The first clinical clue in children often is delayed attainment of one or more developmental milestones.2,3 On average, boys with fragile X syndrome sit without support at 10 months of age and walk and talk at 20 months.2 With few exceptions, affected males have mental retardation, generally of moderate degree. About one third of affected females have mild to severe mental retardation.2 There is a specific pattern of deficits in abstract reasoning, sequential processing, and mathematics. Clinical findings during early childhood may include macrocephaly and frontal bossing (unusually prominent forehead). After puberty, macro-orchidism is present in affected men. Additional findings may include strabismus and mild connective tissue dysplasia, such as mitral valve prolapse, hyperextensible joints, and pes planus. Behavior is characterized by attention deficits, hand flapping, hand biting, and gaze aversion. Family physicians are most likely to encounter the undiagnosed child before school age, when formal testing can confirm cognitive deficits. However, the average age of diagnosis currently is eight years, reflecting the subtlety of features in young children.4
FXTAS is a neurodegenerative disorder with progressive intention tremor and cerebellar ataxia.5 Affected persons present with parkinsonism, peripheral neuropathies, and dementia after age 50 years.
Premature ovarian failure may occur as an isolated clinical finding in women with premutations.1,6 Follicle-stimulating hormone (FSH) levels are elevated in these women even before the onset of premature ovarian failure. Approximately 1 percent of women in the general population have premature ovarian failure, but the prevalence in women with a premutation is 30 times higher.6 Women who are infertile and have prematurely elevated FSH levels should be considered for carrier status testing of the FMR1 premutation. A family history of FXTAS or premature ovarian failure in a child with cognitive deficits is another indicator to initiate genetic testing for fragile X syndrome (Figure 2).
Figure 2.
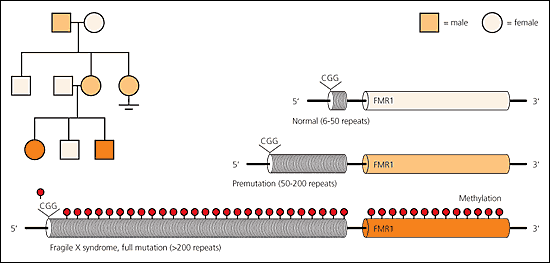
Pedigree of a family with fragile X syndrome. The man in the first generation developed ataxia and tremor in his 60s. He has one son, who is unaffected, and two daughters, one of whom is unaffected and one of whom had menopause in her early 30s and is childless. The unaffected daughter has three children: one unaffected son, and one daughter and one son with fragile X syndrome.
Diagnosis
The diagnosis of fragile X syndrome is confirmed by molecular genetic testing of the FMR1 gene. Prenatal testing is available. FMR1 is characterized by a repetitive CGG trinucleotide sequence, which is repeated six to 50 times in unaffected persons (Figure 2). A full mutation consists of more than 200 CGG repeats in the FMR1 gene, plus hypermethylation, which leads to an inability to produce the FMR1 protein. Almost all males and more than one half of females with full mutations have fragile X syndrome.2 Premutation carriers, who have between 50 and 200 CGG repeats, are not cognitively affected but may have physical or psychiatric findings. In addition, they are susceptible to developing premature ovarian failure and FXTAS. Rarely, the fragile X phenotype occurs in a premutation carrier if hypermethylation is present. Conversely, the phenotype may be absent in a person with a full mutation without hypermethylation, confirming that fragile X syndrome results from the absence of FMR1 protein.
Genetic Counseling and Inheritance
Fragile X syndrome is an X-linked inherited disorder. It is important to diagnose affected patients as early as possible to provide early intervention and supportive care (i.e., specific developmental therapy and an individualized education plan) and to inform parents for further family planning. One half of families in a 2002 survey4 reported having an additional child with fragile X syndrome before the older affected child was diagnosed. Family history collection should include questions about other family members, with particular attention to developmental delay, mental retardation, and psychiatric disorders. In addition, a family history of women with premature ovarian failure and men with FXTAS should be ascertained. A positive family history in a proband with developmental delay should prompt consideration of genetic testing of the FMR1 gene. The American College of Medical Genetics7 recommends testing, regardless of family history, for all males and females with mental retardation of unknown etiology.
Management
Treatment is supportive, requiring a multidisciplinary team and including anxiety-reducing measures, behavior modification, and medications to manage associated psychiatric disorders. Individual education plans are necessary for school-age children.
Resources
Additional information about the diagnosis and management of fragile X syndrome is available at the following Web sites: National Fragile X Foundation (http://www.FragileX.org); GeneTests (http://www.genetests.org); and the American College of Medical Genetics (www.acmg.net/resources/policies/pol-014.asp).
