Arrhythmogenic right ventricular dysplasia (ARVD) is a disorder in which normal myocardium is replaced by fibrofatty tissue. This disorder usually involves the right ventricle, but the left ventricle and septum also may be affected. Although the exact prevalence of ARVD is unknown, it is thought to occur in six per 10,000 persons in certain populations. After hypertrophic heart disease, it is the number one cause of sudden cardiac death in young persons, especially athletes. Patients with ARVD are usually men younger than 35 years who complain of chest pain or rapid heart rate. In some cases, sudden cardiac death is the first presentation. The initial diagnosis of ARVD is based on the presence of major and minor criteria established in 1994. Further confirmation of the diagnosis includes noninvasive studies, such as echocardiography and magnetic resonance imaging of the heart, and invasive studies such as ventricular angiography and endomyocardial biopsy. Patients with ARVD are treated initially with antiarrhythmic agents with serious consideration for automatic implantable cardioverter-defibrillator placement. In patients with persistent symptomatic arrhythmias, radiofrequency ablation, ventriculotomy, or even cardiac transplant may be necessary.
Arrhythmogenic right ventricular dysplasia (ARVD), which was first described in 1977, is a poorly understood yet potentially lethal cause of cardiac disease.1,2 Once thought to be rare, ARVD has been shown to have an incidence of six per 10,000 persons in certain populations.3 In certain Mediterranean and southern U.S. populations, the incidence is as high as 44 per 10,000 persons.3 ARVD accounts for 3 to 4 percent of deaths in sports and 5 percent of sudden cardiac deaths in persons younger than 65 years.4–7 Given the relative frequency of this disorder and its potential for disastrous outcomes, it is important for primary care physicians to be familiar with its presentation, diagnosis, and management.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Although the best imaging technique is debatable, magnetic resonance imaging of the heart can provide noninvasive localization of structural changes and regional dysfunction. | C | 27–30 |
| Electrophysiologic studies can be used to distinguish between idiopathic right ventricular arrhythmias and ARVD. | C | 17,31,32 |
| For chronic management of ARVD, treatment with sotalol (Betapace), beta blockers, propafenone (Rythmol), and amiodarone (Cordarone), alone or in combination, can be used with variable success. | B | 33–35 |
| Placement of an automatic implantable cardioverter-defibrillator should be considered strongly in patients with drug-refractory arrhythmias. | B | 10,37 |
ARVD = arrhythmogenic right ventricular dysplasia.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, see page 1313 orhttps://www.aafp.org/afpsort.xml.
Illustrative Case
A 32-year-old man presented with severe chest pain following exertion. He experienced dizziness, dyspnea, and a tingling sensation in his left arm with the chest pain. He waited for the symptoms to subside but went to the emergency department when the pain had not resolved after 60 minutes. Multiple medical regimens, including lidocaine (Xylocaine) and verapamil (Calan), failed to control his heart rate, and at one point, he required cardioversion because of unstable ventricular tachycardia (VT). His rhythm was finally stabilized using propranolol (Inderal). His event electrocardiography demonstrated no ST-segment changes, but multiple upright premature ventricular contractions were noted in leads II and III and the aVF. Also of note on electrocardiography were negative premature ventricular contractions in leads V1 through V3.
A preliminary diagnosis of right ventricular outflow tract VT was made, and the patient was transferred for cardiac catheterization, cardiac electrophysiologic studies, and magnetic resonance imaging (MRI) of the heart. Results of those studies demonstrated right ventricular dysplasia with a small right lateral wall ventricular aneurysm and inducible sustained monomorphic VT. The diagnosis of ARVD was made, and the patient agreed to receive an automatic implantable cardioverter-defibrillator (AICD) to control future tachycardic events. After AICD implantation, he was discharged on sotalol (Betapace).
Less than a month later, he experienced multiple episodes of sustained VT that were terminated promptly by the AICD. He was readmitted for heart rate control using a combination of intravenous lidocaine and amiodarone (Cordarone) and oral propranolol. Subsequent radiofrequency mapping and ablation, with more than eight arrhythmogenic sites identified and treated in the right ventricle, was successful, and he was discharged on sotalol with no further symptomatic tachycardic events noted.
Pathophysiology
ARVD is characterized by progressive replacement of the right ventricular myocardium by fibrofatty tissue (Figure 1).8,9 The most common location for this tissue transformation is between the anterior infundibulum, right ventricular apex, and inferior or diaphragmatic aspect of the right ventricle, the so-called “triangle of dysplasia” (Figure 2).2 Dysplasia in this region usually leads to dilatations or aneurysms having paradoxical systolic motion (expansion with systole instead of contraction). On the cellular level, there is transmural loss of myocardium in the right ventricular free wall. The left ventricle and septum usually are spared from the fibrofatty transformation, although they may be involved in more extensive cases.10 In addition, the specialized conduction system of the heart usually is spared. The presence of arrhythmias and characteristic findings on electrocardiography (see Clinical Findings and Diagnosis) can be explained by the dispersal of electrically conducting myocytes that can incite tachycardic events as the dysplastic event progresses.11,12
Figure 1.
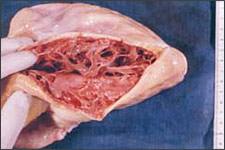
Postmortem heart in a patient with arrhythmogenic right ventricular dysplasia. Note how the right ventricular myocardium and endomyocardium have been replaced by fat.
Figure 2.

Triangle of dysplasia. (RA = right atrium; RV = right ventricle; LV = left ventricle.)
ARVD appears to have a strong familial association, with more than 30 percent of patients reporting a family history of the disorder. Most of these cases demonstrate an autosomal dominant pattern.3,13 Mutations in genes at three loci have been linked to the presence of ARVD.3,5,14 To date, no specific genetic testing for ARVD is available.
Clinical Findings and Diagnosis
Patients with ARVD may present at any age but are usually young or middle-aged (33 ± 14 years); the majority of patients are men (3:1 ratio).10,15 Because of the progressive nature of the disorder, patients may present with any number of symptoms. Palpitations, fatigue, and syncope appear to be the most common symptoms, but patients may have other nonspecific complaints such as abdominal pain and mental confusion (Table 1). In some cases, cardiac arrest following physical exertion such as participation in sports may be the initial presenting problem.8,16
TABLE 1 Signs and Symptoms of ARVD
| Symptoms |
| Abdominal pain |
| Decreased exercise tolerance |
| Dizziness |
| Dyspnea (especially with exertion) |
| Fatigue |
| Mental confusion |
| Palpitations |
| Syncope or fainting |
| Signs |
| Cardiac arrest |
| Peripheral edema |
| Sudden death |
| Tachycardia |
ARVD = arrhythmogenic right ventricular dysplasia.
Screening for ARVD begins with a thorough personal and family history, ideally of first- and second-degree relatives. A personal history of palpitations, especially in a young person, or a family history of sudden cardiac death or death at an early age should raise the index of suspicion for ARVD.
The physical examination is normal in at least 50 percent of patients with ARVD. One important diagnostic clue, if present, is a widely split S2.15 An S3 or S4 heart sound may be noted. Rarely, a murmur may be appreciated. If the right ventricle is greatly dilated, asymmetry of the chest wall may be seen.
The diagnosis of ARVD often is made following a work-up for tachycardia in an otherwise healthy adult. Fifty to 90 percent of persons with ARVD will have characteristic findings on a resting electrocardiogram.8,15,17 These findings include T-wave inversion in the anterior precordial leads (V1 through V6), epsilon waves, or VT with a left bundle branch block pattern, although polymorphic and right bundle branch block patterns also have been reported.9,15,18 Epsilon waves are small deflections just beyond the QRS complex (Figure 3); they are best visualized on a signal averaged electrocardiogram in leads V1 through V3. Any potential in leads V1 through V3 that exceeds the QRS duration in lead V6 by more than 25 milliseconds should be considered an epsilon wave.11
Figure 3.
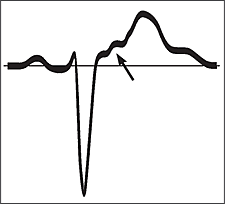
Epsilon wave.
In 1994, a scoring system based on major and minor criteria was implemented to help establish the diagnosis of ARVD (Table 2).4,19,20 Although these criteria are specific, they lack sensitivity and have never been validated, in part because there is no single definitive means of making the diagnosis.3,20,21 The preferred method for making the diagnosis is based on histologic evidence of fibrofatty myocardium. Unfortunately, biopsy lacks sufficient sensitivity because of the segmental nature of the disease process. Once ARVD is suspected, early consultation with a cardiologist is recommended so that further testing may be performed to exclude other diagnoses (Table 3).22,23 These tests involve invasive and noninvasive modalities. Noninvasive testing includes Holter monitoring, exercise stress testing, chest radiography, and MRI of the heart. Invasive testing includes right ventricular angiography, contrast echocardiography, electrophysiologic studies, and endomyocardial biopsy.
TABLE 2 Criteria for the Diagnosis of ARVD*
| Global and/or regional dysfunction and structural alterations | |||
| Major | |||
| Severe dilatation and reduction of right ventricular ejection fraction with no (or only mild) left ventricular impairment | |||
| Localized right ventricular aneurysms | |||
| Severe segmental dilatation of the right ventricle | |||
| Minor | |||
| Mild global right ventricular dilatation and/or ejection fraction with a normal left ventricle | |||
| Mild segmental dilatation of the right ventricle | |||
| Regional right ventricular hypokinesis | |||
| Tissue characterization of the walls | |||
| Major | |||
| Fibrofatty replacement of myocardium on endomyocardial biopsy | |||
| ECG repolarization abnormalities | |||
| Minor | |||
| Inverted T waves in the right precordial leads (V2 and V3) in patients older than 12 years and in the absence of right bundle branch block | |||
| ECG depolarization/conduction abnormalities | |||
| Major | |||
| Epsilon waves or localized prolongation (greater than 110 milliseconds) of the QRS complex in right precordial leads (V1 through V3) | |||
| Minor | |||
| Late potentials visible on signal-averaged ECG | |||
| Arrhythmias | |||
| Minor | |||
| Sustained or nonsustained left bundle branch block type VT documented on ECG, Holter monitoring, or during exercise stress testing | |||
| Frequent ventricular extrasystoles (more than 1,000 per 24 hours on Holter monitoring) | |||
| Family history | |||
| Major | |||
| Familial disease confirmed at autopsy or surgery | |||
| Minor | |||
| Family history of premature sudden death (younger than 35 years) caused by suspected AVRD/DC | |||
| Family history (clinical diagnosis based on present criteria) | |||
ARVD = arrhythmogenic right ventricular dysplasia; ECG = electrocardiography; VT = ventricular tachycardia; ARVD/C = arrhythmogenic right ventricular dysplasia/cardiomyopathy.
*—The diagnosis of ARVD is made if one of the following is met: two major criteria, one major and two minor criteria, or four minor criteria.
TABLE 3 Differential Diagnosis of ARVD
| Anatomic |
| Atrial septal defect |
| Biventricular dysplasia |
| Isolated myocarditis |
| Naxos disease (ARVD associated with palmoplantar keratosis) |
| Right ventricular infarct |
| Right-sided valve insufficiency |
| Uhl’s anomaly (congenital absence of right ventricular myocardium) |
| Arrhythmias |
| Benign extrasystoles |
| Bundle branch reentry |
| Dilated cardiomyopathy VT |
| Idiopathic right ventricular arrhythmia |
| Ischemic heart disease VT |
| Right ventricular outflow tract VT |
| Supraventricular tachycardia |
ARVD = arrhythmogenic right ventricular dysplasia; VT = ventricular tachycardia.
Results of Holter monitoring and exercise stress testing usually are normal. In more advanced cases of ARVD, stress testing may induce VT.15 Chest radiographs usually are normal unless extensive ventricular enlargement is present.
It is debatable which imaging modality is the best for diagnosing ARVD.24 Contrast echocardiography and right ventricular angiography are able to identify ventricular aneurysms and areas of dyskinesis in the triangle of dysplasia but require invasive techniques.25,26 Heart MRI is able to provide noninvasive localization of structural changes and regional dysfunction, although identification of intramyocardial fibrofatty change is difficult.27–30
Electrophysiologic studies are used to detect delayed potentials that can lead to tachycardic events. With these studies, it is possible to determine the number of arrhythmic morphologies, the ease of tachycardia induction, tolerance to tachycardic events, and the capacity for the tachycardia to develop into lethal arrhythmias. Electrophysiologic studies also can be used to distinguish between idiopathic right ventricular arrhythmias, which tend to have a relatively benign course, and ARVD.17,31,32
Endomyocardial biopsy has been considered the preferred method for diagnosis of ARVD. It has a specificity of 92 percent. However, its sensitivity is low (less than 20 percent) because tissue sampling is taken from the septum, where the risk of perforation is low and the likelihood of active disease is minimal.31
Treatment
One of the major goals when treating ARVD is to prevent sudden cardiac death. Treatment decisions should be made in conjunction with a cardiologist or cardiac electrophysiologist. Although there is no way to cure ARVD, the arrhythmias can be managed. Available therapies include lifestyle modifications, antiarrhythmic medications, radiofrequency ablation, defibrillator implantation, and other surgery (Figure 4).
The first step in management is to educate patients about the disease. Although no definitive evidence exists regarding lifestyle modifications, patients should avoid activities that may trigger a tachycardic event, such as excessive physical exertion. Once effective control of arrhythmias is achieved, physical activities may be increased under close medical supervision.
Figure 4. Diagnosis and Management of ARVD
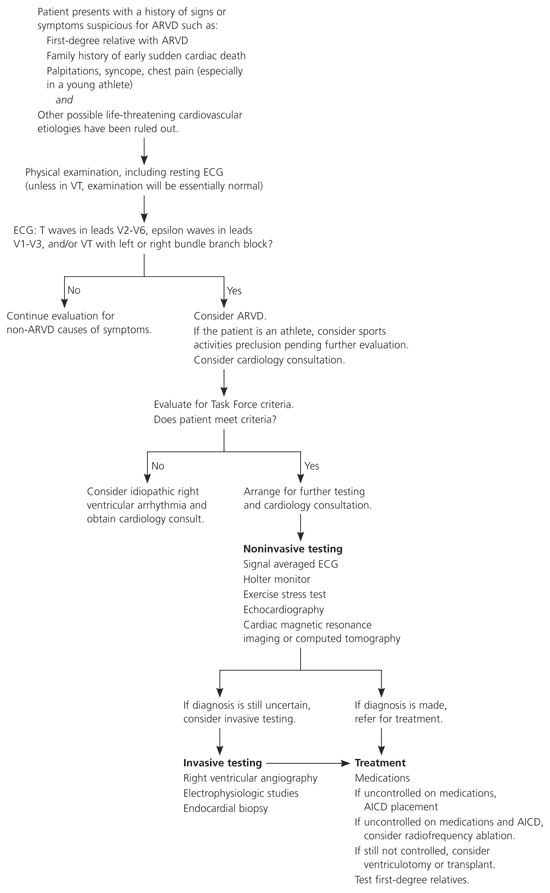
Algorithm for the diagnosis and management of ARVD. (ARVD = arrhythmogenic right ventricular dysplasia; ECG = electrocardiography; VT = ventricular tachycardia; AICD = automatic implantable cardioverter-defibrillator.)
Antiarrhythmic medications are the initial and most commonly used therapy in ARVD.33 No single drug has been shown to be completely effective in controlling or abolishing the arrhythmias, although intravenous amiodarone reportedly has been effective in terminating acute VT in patients with ARVD.8,10 Sotalol and amiodarone are class III drugs. Other drug regimens include class II antiarrhythmics (beta blockers such as propranolol), alone or in combination with class Ia drugs (procainamide [Pronestyl]) or class Ic drugs (flecainide [Tambocor], propafenone [Rythmol]), class Ic drugs alone, and amiodarone with class Ic or class II drugs. For chronic management of ARVD, treatment with sotalol, beta blockers, propafenone, and amiodarone alone or in combination is used most often with variable success.34,35
Radiofrequency ablation is used in cases of drug refractory or incessant VT, frequent tachycardia following internal defibrillator placement, and localized arrhythmia sites.10,11 The goal of radiofrequency ablation is to eliminate conduction pathways that are critical to the perpetuation of arrhythmias. Because radiofrequency ablation is completely successful in only 30 to 65 percent of cases, many patients will require more than one ablation session. These so-called relapses are often the result of disease progression that creates a new reentrant circuit.
The AICD works by providing antitachycardia pacing and defibrillation shocks as needed when arrhythmias occur. As with optimal antiarrhythmic treatment, the timing of AICD placement is debatable because no reliable risk stratification exists for patients with ARVD.36 In general, placement of an AICD should be considered strongly in patients with drug-refractory arrhythmias.10,37 Other indications for an AICD include younger age at onset, cardiac arrest, and left ventricular involvement (Table 4).37–39 The most significant relative contraindication to AICD placement is incessant VT.37 Other contraindications are listed in Table 4.37–39 Complications of AICD placement are rare but include lead perforation, lead failure, infection, and perioperative mortality.38
TABLE 4 Relative Indications and Contraindications for AICD Placement in ARVD
| Indications |
| Sudden cardiac death or cardiac arrest |
| Drug-refractory VT |
| VF/VT with hemodynamic compromise |
| Younger age of onset (less than 35 years) |
| Left ventricular involvement |
| Medication intolerance/noncompliance |
| Contraindications |
| Frequent tachyarrhythmias (i.e., sustained VF/VT) not responsive to pacing or anti-arrhythmic medications |
| VF/VT with a reversible trigger |
| Terminal illness |
| Life expectancy of less than six months |
AICD = automatic implantable cardioverter-defibrillator; ARVD = arrhythmogenic right ventricular dysplasia; VF = ventricular fibrillation; VT = ventricular tachycardia.
If the above treatments fail, ventriculotomy and transplantation are options for management. Right ventriculotomy is performed by disconnecting the right ventricle from the rest of the heart. Heart transplantation remains a final option for patients with bilateral ventricular involvement and uncontrollable hemodynamic compromise.40
Prognosis and Course
ARVD is a progressive disease. The fact that it may be found as the cause of sudden cardiac death in young athletes, or as an incidental finding at autopsy in older adults, illustrates the unpredictable course of this disease. Although multiple studies have demonstrated a favorable long-term prognosis in properly treated patients, few long-term prospective studies have been performed to determine if the course of ARVD can be modified.41 Therefore, lifelong monitoring of patients with ARVD by the primary care physician-cardiologist team is recommended.
