Cardiomyopathy is an anatomic and pathologic diagnosis associated with muscle or electrical dysfunction of the heart. Cardiomyopathies represent a heterogeneous group of diseases that often lead to progressive heart failure with significant morbidity and mortality. Cardiomyopathies may be primary (i.e., genetic, mixed, or acquired) or secondary (e.g., infiltrative, toxic, inflammatory). Major types include dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, and arrhythmogenic right ventricular cardiomyopathy. Although cardiomyopathy is asymptomatic in the early stages, symptoms are the same as those characteristically seen in any type of heart failure and may include shortness of breath, fatigue, cough, orthopnea, paroxysmal nocturnal dyspnea, and edema. Diagnostic studies include B-type natriuretic peptide levels, baseline serum chemistries, electrocardiography, and echocardiography. Treatment is targeted at relieving the symptoms of heart failure and reducing rates of heart failure–related hospitalization and mortality. Treatment options include pharmacotherapy, implantable cardioverter-defibrillators, cardiac resynchronization therapy, and heart transplantation. Recommended lifestyle changes include restricting alcohol consumption, losing weight, exercising, quitting smoking, and eating a low-sodium diet.
Cardiomyopathy is an anatomic and pathologic diagnosis associated with muscle or electrical dysfunction of the heart. The American Heart Association (AHA) defines cardiomyopathy as a heterogeneous group of diseases of the myocardium, usually with inappropriate ventricular hypertrophy or dilatation.1 There are various causes of cardiomyopathy, most of which are genetic. Cardiomyopathy may be confined to the heart or may be part of a generalized systemic disorder, often leading to cardiovascular death or progressive heart failure–related disability.1
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Heart failure should be managed in accordance with the 2005 American College of Cardiology/American Heart Association guidelines. | C | 14 |
| Cardiac resynchronization therapy should be considered in patients with New York Heart Association class III or IV heart failure who remain symptomatic despite optimal pharmacologic therapy. | B | 5, 14 |
| An implantable cardioverter-defibrillator should be placed in patients with cardiomyopathy who are at risk of sudden death. | B | 1 |
| Heart transplantation should be considered in adults with cardiomyopathy who are refractory to maximal medical therapy. | B | 2, 3, 17, 33 |
| Heart transplantation is the treatment of choice in children with idiopathic restrictive cardiomyopathy. | B | 9 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
Epidemiology
In 2006, the AHA classified cardiomyopathies as primary (i.e., genetic, mixed, or acquired) or secondary (e.g., infiltrative, toxic, inflammatory).1 The four major types are dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, and arrhythmogenic right ventricular cardiomyopathy (Table 11–9 ).
Table 1 Diagnostic and Treatment Considerations for Cardiomyopathies
| Type* | Signs and symptoms | Diagnostic considerations | Treatment considerations |
|---|---|---|---|
| Dilated cardiomyopathy | Shortness of breath, fatigue, cough, orthopnea, paroxysmal nocturnal dyspnea, edema | ECG shows LVH Echocardiography shows enlarged ventricular chamber, normal or decreased wall thickness, systolic dysfunction | Pharmacologic therapy based on the 2005 ACC/AHA heart failure guidelines (see Figure 1), cardiac resynchronization therapy, implantable cardioverter-defibrillator, surgical revascularization, left ventricular assist device, salt restriction, smoking cessation, cardiac rehabilitation |
| Hypertrophic cardiomyopathy | Same as dilated cardiomyopathy; sudden cardiac death | ECG shows LVH, large QRS complex, Q-waves, and frequent T-wave inversion Echocardiography shows LVH of unknown etiology with reduction in ventricular chamber volume | Pharmacologic therapy based on the 2005 ACC/AHA heart failure guidelines (see Figure 1), septal myomectomy (only in patients with obstructive hypertrophic cardiomyopathy), biventricular pacing, septal alcohol ablation, implantable cardioverter-defibrillator |
| Restrictive cardiomyopathy | Pulmonary congestion, dyspnea on exertion, decreased cardiac output, syncope | ECG shows LVH Echocardiography shows biatrial enlargement, normal or reduced ventricular volume, normal left ventricle wall thickness, normal systolic function, impaired ventricular filling | Chelation therapy, phlebotomy, bone marrow transplantation, salt restriction, implantable cardioverter-defibrillator, cardiac transplantation (in children) |
| Arrhythmogenic right ventricular cardiomyopathy | Syncope, atypical chest pain, initial episode of ventricular tachycardia, recurrent ventricular tachycardia | ECG shows abnormal repolarization, small-amplitude potentials at end of QRS complex (epsilon wave) Echocardiography shows segmental wall abnormalities, with or without wall motion abnormalities Electrophysiology testing, cardiac magnetic resonance imaging | Beta blockers, antiarrhythmics, catheter ablation, implantable cardioverter-defibrillator, cardiac transplantation |
*— Listed from most to least common.
ACC = American College of Cardiology; AHA = American Heart Association; ECG = electrocardiography; LVH = left ventricular hypertrophy.
Dilated cardiomyopathy, the most common form, affects five in 100,000 adults and 0.57 in 100,000 children.10,11 It is the third leading cause of heart failure in the United States behind coronary artery disease (CAD) and hypertension.1 Hypertrophic cardiomyopathy, the leading cause of sudden death in athletes, is an autosomal dominant disease with an incidence of one in 500 persons.1,12 Restrictive cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy are rare, and their diagnoses require a high index of suspicion.
Etiology
The causes of cardiomyopathies are varied (Table 2).1 Dilated cardiomyopathy in adults is most commonly caused by CAD (ischemic cardiomyopathy) and hypertension, although viral myocarditis, valvular disease, and genetic predisposition may also play a role.1,13,14 In children, idiopathic myocarditis and neuromuscular diseases are the most common etiologies of dilated cardiomyopathy, and generally occur during the first year of life.3 Neuromuscular diseases that may cause dilated cardiomyopathy in children include Duchenne muscular dystrophy; Becker muscular dystrophy; and Barth syndrome, which is an X-linked genetic disorder consisting of dilated cardiomyopathy, skeletal myopathy, and neutropenia.1,15
Table 2 Causes of Cardiomyopathy
| Primary | |
| Genetic | |
| Arrhythmogenic right ventricular cardiomyopathy | |
| Hypertrophic cardiomyopathy | |
| Mixed (genetic and nongenetic) | |
| Dilated cardiomyopathy | |
| Restrictive cardiomyopathy | |
| Acquired | |
| Myocarditis (inflammatory cardiomyopathy) | |
| Peripartum (or postpartum) cardiomyopathy | |
| Stress cardiomyopathy | |
| Secondary | |
| Autoimmune (systemic lupus) | |
| Electrolyte imbalance | |
| Endocrine (diabetes, hypothyroidism) | |
| Endomyocardial (fibrosis) | |
| Infiltrative (amyloidosis, Gaucher disease) | |
| Inflammatory (sarcoidosis) | |
| Neurologic (neurofibromatosis) | |
| Nutritional (beriberi) | |
| Radiation | |
| Storage (hemochromatosis) | |
| Toxic (medications) | |
| Velocardiofacial syndrome | |
Information from reference 1.
Hypertrophic cardiomyopathy is caused by 11 mutant genes with more than 500 individual transmutations.16 The most common variation involves the beta-myosin heavy chain and myosin-binding protein C.1,17 Not all persons with a hypertrophic cardiomyopathy genetic defect are symptomatic. This is most likely because of the phenotypic diversity of hypertrophic cardiomyopathy, and not the consequence of environmental impact or additional genetic modifiers.1
Restrictive cardiomyopathy is an uncommon form that occurs when the ventricles become too stiff to contract. This is often the result of an infiltrative process, such as sarcoidosis, hemochromatosis, amyloidosis, and abnormalities related to desmin (a protein marker found in sarcomeres).1,18,19 One of the familial forms of restrictive cardiomyopathy has a troponin mutation that is the basis of restrictive and hypertrophic cardiomyopathy.1
Arrhythmogenic right ventricular cardiomyopathy is an autosomal dominant, inherited disorder of the muscle of the right ventricle. It may lead to syncope, ventricular arrhythmias, heart failure (less common), or sudden death.1,2 In arrhythmogenic right ventricular cardiomyopathy, the myocardium is replaced by fatty and fibrous tissue. This causes pathologic changes that lead to cardiac compromise.3 The same infiltrative process may also affect the left ventricle.1
Family physicians may also encounter peripartum (or postpartum) cardiomyopathy and alcohol-related cardiomyopathy.1 Peripartum cardiomyopathy is a rare dilated cardiomyopathy with onset in the third trimester of pregnancy or in the first five months postpartum. It tends to occur in multiparous women older than 30 years who are obese and have had preeclampsia. Alcoholism may also lead to a dilated cardiomyopathy that is potentially reversible with abstinence from alcohol use.
Clinical Presentation
Although cardiomyopathies may be asymptomatic in the early stages, most symptoms are typical of those seen in any type of heart failure, whether systolic (reduced ejection fraction) or diastolic (preserved ejection fraction).
Symptoms of heart failure may include shortness of breath, fatigue, cough, orthopnea, paroxysmal nocturnal dyspnea, and edema. This presentation is common in patients with dilated cardiomyopathy. Although the life expectancy of patients with cardiomyopathy varies by etiology, the mortality rate is 20 percent at one year and 70 to 80 percent at eight years for most patients who develop heart failure.12
Patients with hypertrophic cardiomyopathy may present with heart failure, although sudden cardiac death may be the initial presentation.17 Most patients with hypertrophic cardiomyopathy have a propensity to develop dynamic obstruction produced by anterior motion of the mitral valve.
Restrictive cardiomyopathy typically leads to diastolic heart failure from poor filling during diastole and classic heart failure symptoms (e.g., pulmonary congestion, dyspnea on exertion, decreased cardiac output) that progress as systolic dysfunction increases. However, syncope may occur, and sudden death is rare.4
In arrhythmogenic right ventricular cardiomyopathy, symptoms of heart failure are uncommon. Syncope, atypical chest pain, an initial episode of ventricular tachycardia, and recurrent ventricular tachycardia are the primary symptoms.3 In addition, the genetic defect of arrhythmogenic right ventricular cardiomyopathy has cutaneous manifestations, such as Naxos disease, which is characterized by woolly (i.e., extreme curly, kinked) hair and palmoplantar keratoderma.1
Diagnostic Evaluation
The most common clinical presentation in patients with cardiomyopathy is heart failure. The evaluation for underlying causes of heart failure includes a thorough history and physical examination with baseline chemistries, including B-type natriuretic peptide (BNP) levels, echocardiography, and electrocardiography (ECG); chest radiography should be performed on initial presentation.14
In response to elevated volume and filling pressures associated with heart failure, the ventricles secrete BNP into the bloodstream.20 This neurohormone, easily measured in plasma, has been shown to be highly sensitive and specific in the diagnosis of heart failure in patients with acute dyspnea.21 One study found that BNP level was the most accurate predictor of heart failure as the cause of acute dyspnea in the emergency setting.22 The mean serum level of BNP was 675 ± 450 pg per mL (675 ± 450 ng per L) in patients with heart failure, compared with 110 ± 225 pg per mL (110 ± 225 ng per L) in patients with non-heart failure etiologies.
The Heart and Soul Study found that BNP measurement is not a useful screening test in asymptomatic patients with known coronary disease.23 Conversely, the Heart Outcomes Prevention Evaluation Study found that BNP measurement provides the best clinical prediction in the secondary prevention population.24 In the ambulatory setting, BNP levels may be useful in distinguishing patients who need urgent evaluation for possible acutely decompensated heart failure from those who are short of breath for other reasons.
Echocardiography is another key diagnostic modality for patients with suspected cardiomyopathy. In dilated cardiomyopathy, echocardiography typically demonstrates an enlarged ventricular chamber with normal or decreased wall thickness and systolic dysfunction.1 The ECG will show left ventricular hypertrophy. In patients with familial idiopathic dilated cardiomyopathy, the American College of Cardiology (ACC)/AHA heart failure guidelines recommend screening asymptomatic first-degree relatives with echocardiography and ECG, as well as possible referral to a cardiovascular genetics center.14
In patients with hypertrophic cardiomyopathy, echocardiography reveals left ventricular hypertrophy of unknown etiology with a reduction in ventricular chamber volume.1 The ECG also demonstrates left ventricular hypertrophy, as well as a large QRS complex, Q-waves with no history of CAD, and frequent T-wave inversion. A harsh murmur heard at the left sternal edge that increases with Valsalva maneuver and the standing position is often heard on auscultation. The ACC and the European Society of Cardiology recommend that first-degree relatives and other family members of patients with hypertrophic cardiomyopathy receive a history and physical examination, ECG, and echocardiography annually between 12 and 18 years of age.17
In patients with restrictive cardiomyopathy, echocardiography tends to show biatrial enlargement with a normal or reduced ventricular volume, normal left ventricle wall thickness, normal systolic function, and impaired ventricular filling.1 The ECG typically reveals decreased voltage despite signs of left ventricular hypertrophy.
Diagnostic evaluation for arrhythmogenic right ventricular cardiomyopathy differs from the other forms of cardiomyopathy. Echocardiography typically reveals global or segmental wall abnormalities with or without wall motion abnormalities.1 The ECG shows abnormal repolarization and small-amplitude potentials at the end of the QRS complex (epsilon wave). The diagnosis is typically made by evaluating for electrical, functional, and anatomic abnormalities that may have been evaluated for previously because of a sudden arrhythmia, syncope, or cardiac arrest.1 Alternatively, cardiac magnetic resonance imaging has been used in patients who have a high pretest probability.
The Athlete's Heart
Athletes, especially those who follow intense training regimens, may develop changes in cardiac structure as a normal physiologic response. Such changes may include eccentric cardiac hypertrophy with a resultant increase in left ventricular volume, and mass or concentric hypertrophy with increased ventricular wall thickness, but no change in cavity size.25 Although these changes are not considered to be pathologic in athletes, underlying conditions (most notably hypertrophic cardiomyopathy) that place them at risk of sudden death may be present. To guide physicians who treat athletes, the AHA issued recommendations for preparticipation cardiovascular screening (Table 3).26 A positive answer on questioning or an abnormal finding should prompt evaluation for a possible underlying cardiac condition.
Table 3 American Heart Association Screening Questions for Preparticipation Cardiovascular Evaluation in Athletes
| Is there a personal history of exertional chest pain or discomfort? |
| Is there a personal history of unexplained syncope or near syncope? |
| Is there a personal history of dyspnea or fatigue with exercise? |
| Is there a personal history of heart murmur? |
| Is there a personal history of elevated blood pressure? |
| Is there a family history of premature cardiac death before 50 years of age? |
| Is there a family history of disabling heart disease before 50 years of age? |
| Is there a family history of conditions known to increase cardiac risk (e.g., dilated or hypertrophic cardiomyopathy)? |
| Evaluate for heart murmur. |
| Evaluate for femoral pulses. |
| Evaluate for physical features suggestive of Marfan syndrome. |
| Obtain blood pressure. |
note:A positive answer on questioning or an abnormal finding should prompt evaluation for a possible underlying cardiac condition.
Adapted from Maron BJ, Thompson PD, Ackerman MJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007;115(12):1646.
Routine ECG, echocardiography, and stress testing are not recommended as part of the preparticipation physical examination.27 However, a recent controversial AHA scientific statement advises physicians to consider ECG in all children who take medications for attention-deficit/hyperactivity disorder, regardless of athletic participation.28
Treatment
Treatment for dilated cardiomyopathy is directed at the underlying disease. Most patients have heart failure; therefore, treatment should follow the ACC/AHA heart failure guidelines (Figure 1).14 Lifestyle changes should include reduced alcohol consumption, weight loss, exercise, smoking cessation, and a low-sodium diet.14 Treatment includes administration of an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a loop diuretic, spironolactone (Aldactone) for New York Heart Association (NYHA) class III or IV heart failure, and a beta blocker. Metoprolol (Lopressor), carvedilol (Coreg), and bisoprolol (Zebeta) are the only beta blockers with proven benefit in heart failure, according to randomized controlled trials.14,29–31
Figure 1. Stages of Heart Failure and Treatment
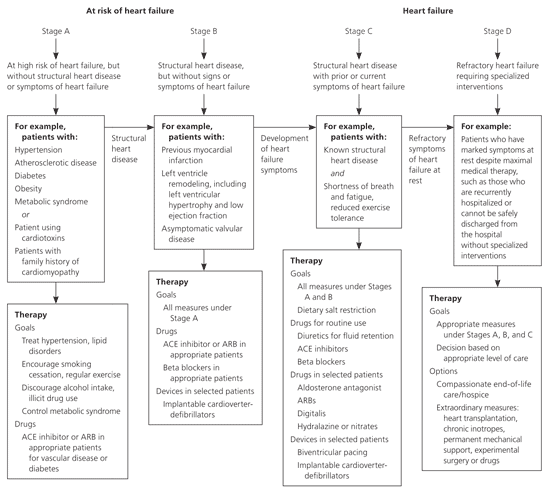
American College of Cardiology/American Heart Association heart failure guidelines. (ACE = angiotensin-converting enzyme; ARB = angiotensin receptor blocker.)
Adapted from Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 Guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society [published correction appears in Circulation. 2006;113(13): e682–e683]. Circulation. 2005;112(12):1830.
The African American Heart Failure Trial demonstrated a significant reduction in hospitalizations and an increase in quality of life with the use of isosorbide dinitrate/hydralazine (Bidil).32 Salt restriction, smoking cessation, and a cardiac rehabilitation program, if indicated, are also important. Diastolic heart failure is typically treated with the same medical regimen as systolic heart failure.
Cardiac resynchronization therapy is a nonpharmacologic option in appropriate patients who have evidence of dyssynchrony and who have NYHA class III or IV heart failure and continued symptoms despite maximal medical therapy.5,14 An implantable cardioverter-defibrillator may be needed for primary or secondary prevention in patients at high risk of sudden death.1 Referral to an electrophysiologist is needed for final determination of eligibility for resynchronization or placement of an implantable cardioverter-defibrillator.5
The Surgical Treatment for Ischemic Heart Failure Trial found that in patients with heart failure caused by CAD, surgical revascularization with surgical ventricular reconstruction does not lead to greater improvement in symptoms or exercise tolerance, or a reduction in death rate, compared with surgical revascularization alone.6 Transplantation may be an option for patients if all other treatments have failed.33 If the patient is ineligible for transplantation, a left ventricular assist device may improve survival and quality of life.7
The management of hypertrophic cardiomyopathy is focused on reducing symptoms and complications from heart failure by following ACC/AHA guidelines. Because many patients with hypertrophic cardiomyopathy have diastolic dysfunction and typically need higher filling pressures, diuretics should be used with caution.17 If patients do not respond to drug therapy, treatment is dictated by whether the patient has nonobstructive hypertrophic cardiomyopathy or obstructive hypertrophic cardiomyopathy. Nonobstructive end-stage disease that is refractive to maximal medical therapy requires heart transplantation17; this represents most patients with hypertrophic cardiomyopathy.34
Patients with obstructive hypertrophic cardiomyopathy may benefit from septal myomectomy, biventricular pacing, or septal alcohol ablation. An implantable cardioverter-defibrillator may be needed in patients at high risk of sudden death.17
The treatment of restrictive cardiomyopathy is difficult because the underlying processes usually do not respond to intervention. Therapies directed at specific forms of this condition include chelation therapy, phlebotomy, bone marrow transplantation, salt restriction, and implantable cardioverter-defibrillator placement.8 In children, restrictive cardiomyopathy is primarily idiopathic, and transplantation is the treatment of choice. This is often required within four years of diagnosis.9
Pharmacologic treatment of patients with arrhythmogenic right ventricular cardiomyopathy is directed at arrhythmia suppression and involves beta blockers, such as sotalol (Betapace), with or without amiodarone (Cordarone).2 Nonpharmacologic options include catheter ablation, implantable cardioverter-defibrillator placement, and cardiac transplantation in patients refractory to rhythm control interventions.2,3
