
Am Fam Physician. 2009;80(11):1261-1268
Patient information: See related handout on von Willebrand disease, written by the authors of this article.
Author disclosure: Nothing to disclose.
Von Willebrand disease is an inherited condition characterized by deficiency of von Willebrand factor, which is essential in hemostasis. The National Heart, Lung, and Blood Institute has released new evidence-based guidelines for the diagnosis and management of the disease. There are three major subtypes of von Willebrand disease, classified as partial quantitative deficiency (low levels) of von Willebrand factor (type 1), qualitative deficiency (type 2), or virtually complete deficiency (type 3). Diagnosis is usually made by reviewing the patient's personal and family history of bleeding and by clinical evaluation for more common reasons for bleeding, supplemented with laboratory tests. Assessment may be used to determine bleeding risk before surgery and other invasive procedures, and to diagnose reasons for unexplained hemorrhaging. Von Willebrand factor levels of 30 IU per dL or lower are required for the definite diagnosis of inherited von Willebrand disease. Persons with levels of 30 to 50 IU per dL may not have the disease, but may need agents to increase von Willebrand factor levels during invasive procedures or childbirth. Treatment is tailored to the subtype of the disease: increasing plasma concentration of von Willebrand factor by releasing endogenous stores with desmopressin or replacing nonexistent or ineffective von Willebrand factor by using human plasma–derived, viral-inactivated concentrates; treatment is often combined with hemostatic agents that have mechanisms other than increasing von Willebrand factor. Regular prophylaxis is seldom required, and treatment is initiated before planned invasive procedures or in response to bleeding.
Von Willebrand disease (VWD) encompasses a group of inherited bleeding disorders related to qualitative or quantitative defects of von Willebrand factor (VWF), which is essential in hemostasis. The disease leads to bleeding from impaired platelet adhesion and aggregation, and may be accompanied by a reduced concentration of factor VIII. Undiagnosed VWD likely accounts for many women with menorrhagia and patients with mild to moderate hemorrhage during or after invasive procedures. In screening assessments of large asymptomatic populations, the prevalence of VWD has been reported to be as high as 1.3 percent. However, only about 0.01 percent of the population had clinically recognized symptoms associated with abnormal VWD test results.1 Of note, average VWF levels are 25 percent lower in persons with type O blood compared with others in the ABO blood group, and many inflammatory conditions and pregnancy increase VWF levels, confounding the diagnosis.2 It is important for family physicians to know when to suspect VWD, to know how to evaluate for the disease, and to understand the principles of management. This article summarizes recent evidence-based guidelines on the diagnosis and management of VWD from the National Heart, Lung, and Blood Institute (NHLBI).3-5
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Evaluation for VWD should be considered preoperatively for currently asymptomatic persons if personal or family history of bleeding is a concern; for persons with current symptoms or a history of increased bleeding, abnormal laboratory study results, or a family history of a bleeding disorder; and for persons with a previous VWD diagnosis, but no supporting laboratory documentation. | C | 1, 3, 4 |
| Clinical history in patients with possible VWD should focus on episodes of excessive bleeding, including spontaneity, severity, cause, sites, and duration of bleeding; and ease with which bleeding was stopped. Patients should also be asked about medication use (e.g., aspirin, clopidogrel [Plavix], heparin, nonsteroidal anti-inflammatory drugs, warfarin [Coumadin]). | C | 1, 3, 4, 12–15 |
| Initial tests for a bleeding disorder rule out more common causes of bleeding. These tests include complete blood and platelet counts, PTT, PT, and possibly fibrinogen level or thrombin time. Initial tests for VWD (VWF:Ag, VWF:RCo, factor VIII) confirm VWD. | C | 3–5 |
| Patients with isolated prolonged PTT or with normal PTT, PT, platelet count, and fibrinogen level in the presence of bleeding signs or symptoms should receive VWF:Ag, VWF:RCo, and factor VIII assays to test for VWD. | C | 1, 3, 4, 7, 16 |
Pathophysiology
Synthesis of VWF occurs in the vascular endothelium and megakaryocytes. It is released from platelets and endothelial cells when they are activated and binds to factor VIII in the circulation, prolonging its half-life6; VWF has a half-life of approximately 12 hours. With vascular injury, VWF bridges between exposed collagen and platelets. Factor VIII is also released from the VWF and facilitates the formation of thrombin and a fibrin clot.6
Classification
There are three major subtypes of VWD (Table 17): partial quantitative VWF deficiency (type 1, 75 percent of patients with VWD), qualitative VWF deficiency (type 2, 25 percent of patients with VWD), and virtually complete VWF deficiency (type 3, rare).8 Type 2 disease is further divided into four variants (2A, 2B, 2M, 2N) on the basis of the phenotype. Although it can be difficult to distinguish the subtypes and separate true VWD from mildly low VWF levels, these distinctions are important in treatment decisions and prognosis.7,9
TYPE 1
In type 1 VWD, the level of plasma VWF is low, but the existing VWF functions normally. Levels of blood clotting factor VIII usually parallel those of VWF and, therefore, may also be reduced. However, a normal factor VIII level does not rule out VWD.
In the general population, the mean level of plasma VWF is 100 IU per dL, with a normal reference range between 50 and 200 IU per dL. The 5 percent of persons with VWF levels of less than 50 IU per dL include those with VWD and those with slightly low, but nondiagnostic, levels. Self-reported, mild bleeding symptoms (e.g., menorrhagia, prolonged nosebleeds) are common in healthy persons, and the association between bleeding symptoms and mildly to moderately low VWF levels may be coincidental.7 This makes diagnosing type 1 VWD difficult.
TYPE 2
Bleeding symptoms in patients with type 2 VWD are often more severe than in patients with type 1. However, like in type 1, symptoms depend on the severity and extent of the bleeding trigger (often trauma or surgery). Because of the multiple variants of type 2 VWD, collaboration with a hematologist who has expertise in hemostasis is helpful for diagnosis and management.
TYPE 3
Type 3 VWD is characterized by undetectable VWF protein and activity. Factor VIII levels usually are very low (1 to 9 IU per dL).
ACQUIRED VON WILLEBRAND SYNDROME
Acquired von Willebrand syndrome (AVWS) is less common than congenital VWD, occurring in fewer than one in 100,000 adults,10 and is typically associated with one of several mechanisms or medical conditions (Table 23,10,11). Laboratory findings in patients with AVWS are similar to those in patients with congenital VWD.10,11 When clinical and laboratory assessments identify VWD in the absence of a personal or family history of congenital VWD, AVWS should be considered and evaluation for contributing disorders should be initiated. Conversely, AVWS should be considered when bleeding occurs in association with one of its causative conditions, and initial VWD testing performed (evaluation for AVWS and congenital VWD is the same).
| Pathophysiology | Associations |
|---|---|
| Antibodies to VWF | Lymphoproliferative diseases; monoclonal gammopathies; autoimmune diseases, such as systemic lupus erythematosus |
| Shear-induced VWF conformational changes leading to increased proteolysis of VWF | Ventricular septal defect, aortic stenosis, hypertrophic obstructive cardiomyopathy, left ventricular assist device, primary pulmonary hypertension |
| Markedly elevated blood platelet count | Essential thrombocythemia, polycythemia vera, myelofibrosis with myeloid metaplasia, other myeloproliferative disorders |
| Removal of VWF from circulation by aberrant binding to tumor cells | Wilms tumor, certain lymphoproliferative or plasma cell proliferative disorders |
| Decreased VWF synthesis | Hypothyroidism |
| Complications from medication use | Ciprofloxacin (Cipro), valproic acid (Depakene), hydroxyethyl starch (no longer available in the United States), griseofulvin |
Diagnosis
Evaluation for VWD (or other bleeding disorders) should be considered for the following patients: (1) currently asymptomatic patients undergoing a surgical or interventional procedure if personal or family history of bleeding is a concern; (2) patients with current symptoms or a history of increased bleeding, abnormal laboratory study results, or a family history of a bleeding disorder; and (3) patients with a previous VWD diagnosis, but no supporting laboratory documentation.1,3 The initial step in the evaluation should focus on key aspects of the patient and family history. History and examination findings determine the need for further laboratory testing (Figure 1).3
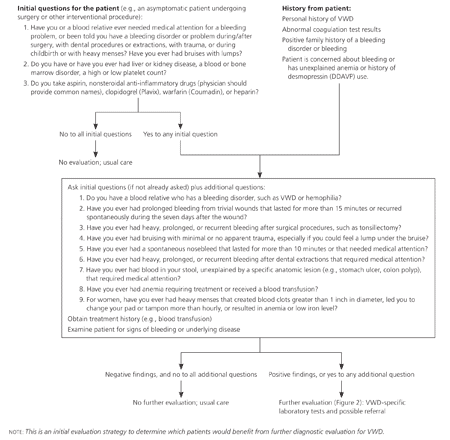
CLINICAL HISTORY AND PHYSICAL EXAMINATION
The history should focus on episodes of excessive bleeding, including spontaneity, severity, cause, sites, and duration of bleeding; type of associated injury; and ease with which bleeding was stopped. A family history of an established bleeding disorder is useful in identifying persons at risk of VWD, but often is not present. Other causes of bleeding (e.g., medications, such as clopidogrel [Plavix], heparin, nonsteroidal anti-inflammatory drugs [NSAIDs], and warfarin [Coumadin]; liver, kidney, blood, or bone marrow diseases) should be considered before the evaluation of VWD continues.1,3,12-15
In patients with VWD, the most common sites of hemorrhage include mucous membranes (usually nosebleeds), skin (bruising), and the uterus in women (menorrhagia, postpartum hemorrhage). Bleeding during or after surgical procedures is possible, and muscle and joint bleeding occur with more severe disease. Most bleeding is mild to moderate and does not require transfusion or prompt a physician visit, making it difficult to separate normal bleeding from that associated with mild VWD.12 Although menorrhagia is common in women, those with VWD are more likely to have more marked menorrhagia with blood clots greater than 1 inch in diameter, bleeding that requires a pad or tampon change more than hourly, and low serum ferritin levels.13
The physical examination should be directed at detecting signs of bleeding, including size, location, and distribution of ecchymoses, hematomas, and petechiae; signs of anemia; and other causes of bleeding, including liver disease, splenomegaly, and gynecologic lesions.
LABORATORY TESTING
No simple, single laboratory test is available to screen for VWD, and the diagnosis is made using several specific laboratory tests. If the history or physical examination suggests a possible bleeding disorder, laboratory testing (Figure 23 ) is appropriate, beginning with evaluation for any hemostasis problem. The initial hemostasis evaluation should include complete blood and platelet counts, partial thromboplastin time, prothrombin time, and possibly fibrinogen level or thrombin time.5,6,8 These tests can exclude other bleeding problems, such as isolated low platelet counts from thrombocytopenic disorders, isolated prolongation of prothrombin time from warfarin use, and low fibrinogen levels from diffuse intravascular coagulation or sepsis. These coagulation factor abnormalities are unlikely to be caused by VWD and require other hematologic assessment. Bleeding time testing is no longer recommended to screen for bleeding disorders.3
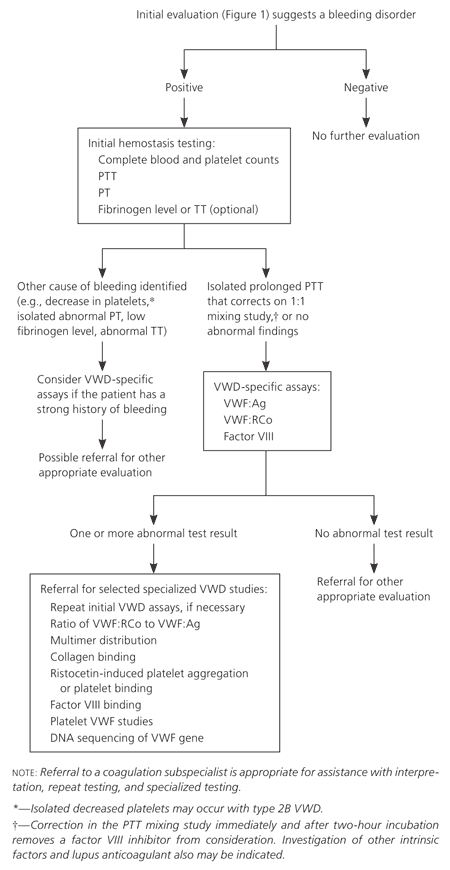
An isolated prolonged partial thromboplastin time or a normal partial thromboplastin time, prothrombin time, platelet count, and fibrinogen level in the presence of signs or symptoms of bleeding suggest the need for VWD-specific testing1,3,7,16 (Figure 23 and Table 33). It is important to note that the initial hemostasis panel will not detect many VWD cases, and patients with a significant personal or family history of bleeding require thorough evaluation even if initial hemostasis findings are normal. In such cases, it may be helpful to consult a coagulation subspecialist. Consultation with a coagulation subspecialist is also appropriate if any of the hemostasis findings are abnormal, to assist in further testing and referral decisions.
| Diagnosis | Initial tests | Additional tests | ||||
|---|---|---|---|---|---|---|
| VWF:RCo (IU per dL)* | VWF:Ag (IU per dL)*† | Factor VIII | Ratio of VWF: RCo to VWF:Ag | RIPA | VWF multimers‡ | |
| Type 1 | < 30 | < 30 | ↓ or normal | > 0.5 to 0.7 | Often normal | Normal |
| Type 2A | < 30 | < 30 to 200 | ↓ or normal | < 0.5 to 0.7 | ↓ | ↓ high-molecular-weight multimers |
| Type 2B | < 30 | < 30 to 200 | ↓ or normal | Usually < 0.5 to 0.7 | ↑ | ↓ high-molecular-weight multimers |
| Type 2M | < 30 | < 30 to 200 | ↓ or normal | < 0.5 to 0.7 | ↓ | Normal |
| Type 2N | 30 to 200 | 30 to 200 | ↓ ↓ | > 0.5 to 0.7 | Normal | Normal |
| Type 3 | < 3 | < 3 | ↓ ↓ ↓ (< 10 IU per dL) | NA | Absent | NA |
| Low VWF | 30 to 50§ | 30 to 50§ | Normal | > 0.5 to 0.7 | Normal | Normal |
| Normal | 50 to 200 | 50 to 200 | Normal | > 0.5 to 0.7 | Normal | Normal |
DIAGNOSING SUBTYPES
Classification of subtypes is based on initial VWD testing and other specialized VWD tests, combined with the clinical history. Separation into types 2 and 3 is usually done in consultation with a coagulation subspecialist.
Diagnosing type 1 VWD and distinguishing it from a low VWF level without VWD can be challenging, especially when VWF levels are only mildly decreased (30 to 50 IU per dL). Therefore, it is important to correlate severity and cause of bleeding with results of laboratory testing.
Testing may need to be repeated because some conditions can alter baseline VWF and factor VIII levels. Stress, such as in a struggling child or anxious adult; very recent exercise; acute or chronic illness; pregnancy; and the use of estrogen or oral contraceptives may falsely elevate VWF and factor VIII levels. Careful processing of blood samples is also important (Table 43).
| Phlebotomy conditions: An atraumatic blood draw limits the exposure of tissue factor from the site and the activation of clotting factors, minimizing falsely high or low values. |
| Patient stress level: Undue stress, such as struggling or crying in children or anxiety in adults, may falsely elevate VWF and factor VIII levels. Very recent exercise may also elevate VWF levels. |
| Additional patient conditions: The presence of an acute or chronic inflammatory illness, pregnancy, or use of estrogen/oral contraceptives may elevate VWF and factor VIII levels. |
| Sample processing: To prevent cryoprecipitation of VWF and other proteins, blood samples for VWF assays should be transported to the laboratory at room temperature. Plasma should be separated from blood cells promptly at room temperature, and the plasma should be centrifuged thoroughly to remove platelets. If plasma samples will be assayed within two hours, they should be kept at room temperature. Frozen plasma samples should be carefully thawed at 98.6° F (37° C) and kept at room temperature for less than two hours before being assayed. |
| Sample storage: Plasma samples that will be stored or transported to a reference laboratory must be frozen promptly at or below −40° F (−40° C) and remain frozen until assayed. |
| Control sample: A control sample that is drawn, processed, stored, and transported under the same conditions as the patient's sample may be helpful in indicating problems in the handling of important test samples. |
Management
There are three main approaches to the treatment of VWD, used individually or in combination. These approaches include increasing plasma concentration of VWF by releasing endogenous VWF stores through stimulation of endothelial cells with desmopressin (DDAVP); replacing VWF by using human plasma–derived, viral-inactivated concentrates; and promoting hemostasis using hemostatic agents with mechanisms other than increasing VWF. Regular prophylaxis is seldom required, and treatment is initiated before planned invasive procedures or in response to bleeding.
DESMOPRESSIN
Desmopressin is a synthetic derivative of the antidiuretic hormone. It is used in type 1 VWD and in some patients with type 2 VWD. Desmopressin is generally administered for short periods (48 to 72 hours), and no more often than at 24- to 48-hour intervals because of tachyphylaxis and adverse effects. However, if a longer duration or shorter intervals are required, the patient should be monitored for fluid and electrolyte problems because desmopressin may lead to symptomatic hyponatremia.8
FACTOR REPLACEMENT THERAPY
Humate-P and Alphanate are currently the only plasma-derived VWF concentrates approved by the U.S. Food and Drug Administration for the treatment of VWD. These products also contain factor VIII, but differ in ratios of VWF to factor VIII and in content of high-molecular-weight multimers of VWF. Cryoprecipitate is no longer recommended for VWF or factor VIII replacement. Treatment duration varies by procedure (Table 53).
| Major surgery (seven to 14 days of treatment) |
| Cardiothoracic |
| Cesarean delivery |
| Craniotomy |
| Hysterectomy |
| Open cholecystectomy |
| Prostatectomy |
| Minor surgery (one to five days of treatment) |
| Biopsy: breast, cervical |
| Central line placement |
| Complicated dental extractions |
| Gingival surgery |
| Laparoscopic procedures |
| Other procedures (if uncomplicated, single treatment) |
| Cardiac catheterization |
| Cataract surgery |
| Endoscopy (without biopsy) |
| Laceration repair |
| Liver biopsy |
| Simple dental extractions |
ANTIFIBRINOLYTICS AND TOPICAL AGENTS
The oral antifibrinolytic agents epsilon-aminocaproic acid (Amicar) and tranexamic acid (oral formulation no longer available in the United States) are very useful for mucous membrane bleeding, such as with dental procedures.17 Topical agents, such as bovine or human thrombin and fibrin sealants, may be used for accessible minor bleeding.17,18
Women with VWD
MENORRHAGIA
Menorrhagia may be the first sign of VWD in women,19 although other causes of menorrhagia must be ruled out first.14 If menorrhagia is caused by VWD and the patient is not trying to get pregnant, combined oral contraceptives are the treatment of choice. The levonorgestrel-releasing intrauterine device (Mirena) is an alternative in appropriate patients.20,21 Desmopressin, antifibrinolytics, or VWF concentrates have been used to manage menorrhagia in women desiring pregnancy; referral to a hemophilia center should be considered in these patients.
PREGNANCY AND CHILDBIRTH
Before or during pregnancy, women with VWD should be referred to a genetic counselor to discuss the inheritance of the disease,23 and to a pediatric hematologist for evaluation of the infant after delivery. Pregnant women with VWD who have factor VIII or VWF ristocetin cofactor (VWF:RCo) levels of less than 50 IU per dL, or a history of severe bleeding should be referred to a perinatal center that has a hemophilia treatment center or a hematologist with expertise in hemostasis. Before invasive procedures or regional anesthesia during labor and delivery, women who have VWD should receive factor VIII and VWF:RCo assays to determine the need for and amount of appropriate prophylaxis.23,24
Desmopressin therapy initiated during labor may cause significant fluid retention, which is aggravated by use of oxytocin (Pitocin), with risk of hyponatremia and seizures. Therefore, women with VWD and factor VIII levels or VWF:RCo of less than 50 IU per dL should receive VWF replacement with VWF concentrates as opposed to desmopressin. After delivery, NSAIDs may decrease platelet function, aggravating bleeding or increasing the risk of postpartum hemorrhage.25 The elevated VWF levels caused by pregnancy return to baseline within seven to 21 days after delivery,26,27 predisposing women with VWD to delayed postpartum hemorrhage at 21 to 28 days.
editor's note: The authors of this article are members of the NHLBI Von Willebrand Disease Expert Panel.