Hereditary hemorrhagic telangiectasia is an uncommon autosomal dominant disease that occurs in approximately one in 5,000 to 8,000 persons. This multisystem disorder can affect the nose, skin, gastrointestinal tract, lungs, liver, and brain. Epistaxis is the most common presenting problem, occurring in 90 percent of affected patients. Approximately 15 to 30 percent of patients with hereditary hemorrhagic telangiectasia will have an arteriovenous malformation in the lungs and more than 10 percent will have one in the brain. The symptoms of hereditary hemorrhagic telangiectasia are often unrecognized. Many patients, even those with affected family members, may go undiagnosed. Hereditary hemorrhagic telangiectasia is a clinical diagnosis that is based on the presence of three of four criteria (i.e., epistaxis, telangiectasias, visceral arteriovenous malformations, or family history of the disease). Screening and treatment recommendations have been created in an attempt to limit the morbidity and mortality associated with this disease. Patients with confirmed or suspected hereditary hemorrhagic telangiectasia should be screened for brain and lung arteriovenous malformations using magnetic resonance imaging of the brain and contrast echocardiography. Pulmonary arteriovenous malformations can be treated with embolization. Patients with a history of pulmonary arteriovenous malformations or those who have not been screened should use antibiotic prophylaxis before dental treatment, endoscopy, or other procedures that could cause bacteremia because of the risk of paradoxical brain embolism or infection.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu disease, is an autosomal dominant disorder of abnormal blood vessel formation. It occurs in approximately one in 5,000 to 8,000 persons.1–3 Recurrent epistaxis is the most common presenting problem; however, some patients present with a catastrophic event (e.g., sudden death, stroke, pulmonary hemorrhage) related to major organ involvement.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
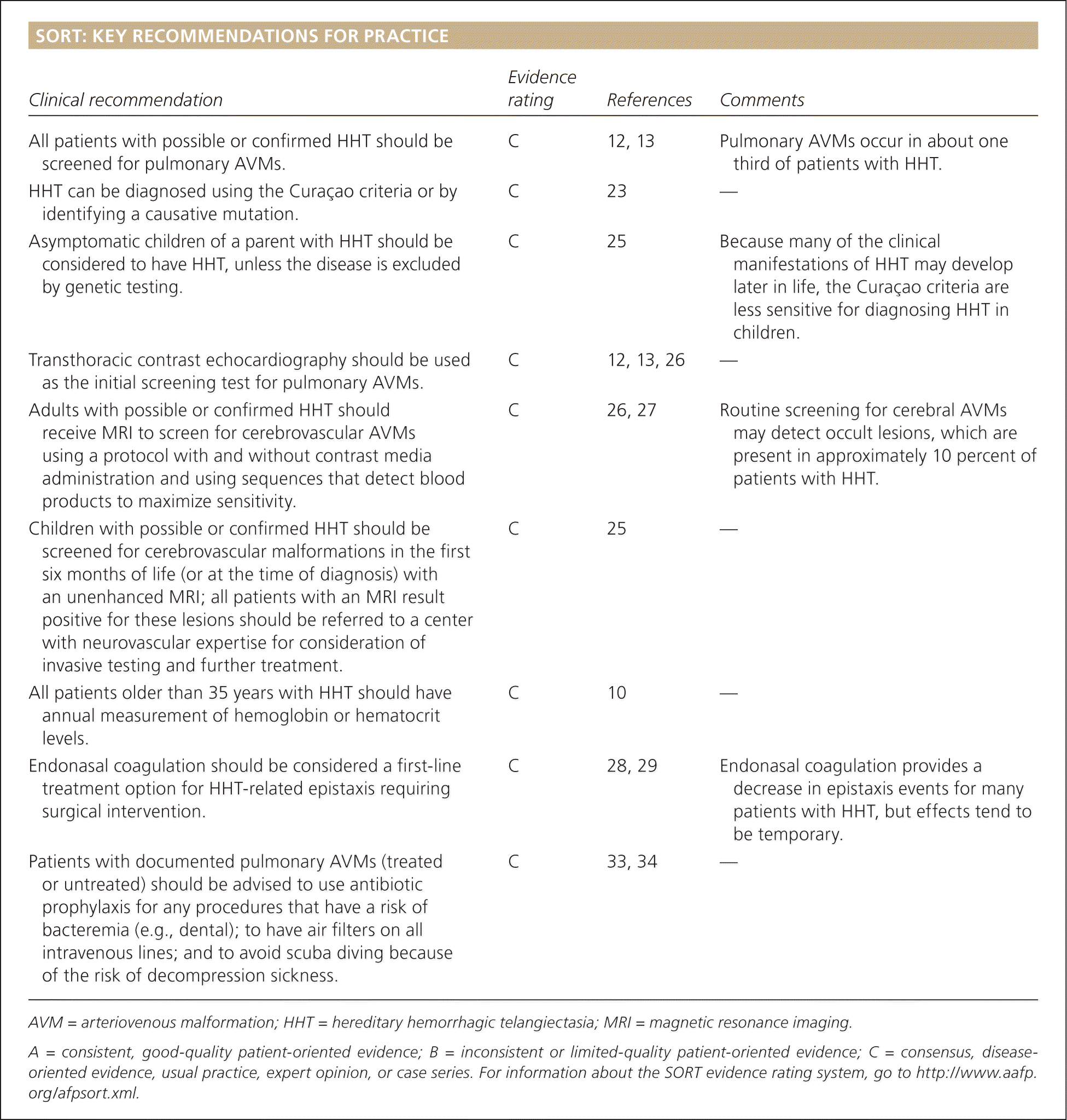
| Clinical recommendation | Evidence rating | References | Comments |
|---|---|---|---|
| All patients with possible or confirmed HHT should be screened for pulmonary AVMs. | C | 12, 13 | Pulmonary AVMs occur in about one third of patients with HHT. |
| HHT can be diagnosed using the Curaçao criteria or by identifying a causative mutation. | C | 23 | — |
| Asymptomatic children of a parent with HHT should be considered to have HHT, unless the disease is excluded by genetic testing. | C | 25 | Because many of the clinical manifestations of HHT may develop later in life, the Curaçao criteria are less sensitive for diagnosing HHT in children. |
| Transthoracic contrast echocardiography should be used as the initial screening test for pulmonary AVMs. | C | 12, 13, 26 | — |
| Adults with possible or confirmed HHT should receive MRI to screen for cerebrovascular AVMs using a protocol with and without contrast media administration and using sequences that detect blood products to maximize sensitivity. | C | 26, 27 | Routine screening for cerebral AVMs may detect occult lesions, which are present in approximately 10 percent of patients with HHT. |
| Children with possible or confirmed HHT should be screened for cerebrovascular malformations in the first six months of life (or at the time of diagnosis) with an unenhanced MRI; all patients with an MRI result positive for these lesions should be referred to a center with neurovascular expertise for consideration of invasive testing and further treatment. | C | 25 | — |
| All patients older than 35 years with HHT should have annual measurement of hemoglobin or hematocrit levels. | C | 10 | — |
| Endonasal coagulation should be considered a first-line treatment option for HHT-related epistaxis requiring surgical intervention. | C | 28, 29 | Endonasal coagulation provides a decrease in epistaxis events for many patients with HHT, but effects tend to be temporary. |
| Patients with documented pulmonary AVMs (treated or untreated) should be advised to use antibiotic prophylaxis for any procedures that have a risk of bacteremia (e.g., dental); to have air filters on all intravenous lines; and to avoid scuba diving because of the risk of decompression sickness. | C | 33, 34 | — |
AVM = arteriovenous malformation; HHT = hereditary hemorrhagic telangiectasia; MRI = magnetic resonance imaging.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
HHT has varying penetrance and expressivity, often leading to delays in or doubts about the diagnosis. The disease is often unrecognized by many physicians; a majority of patients do not know they have it. Most have a mutation in one of two genes that are required for normal angiogenesis.4,5 In addition, vascular endothelial growth factor levels are increased6 and may play a role in abnormal angiogenesis.
A better understanding of the many clinical manifestations of HHT, in addition to better screening protocols and diagnostic testing, has helped to determine that the disease is more common than once thought.
Clinical Features
Patients with HHT have abnormal blood vessel development that manifests as telangiectasias and arteriovenous malformations (AVMs). Both telangiectasias and AVMs represent a direct connection between arteries and veins without bridging capillaries. Telangiectasias occur on mucocutaneous surfaces (i.e., nose, gastrointestinal [GI] tract, and skin); AVMs develop in larger organ systems (i.e., lungs, liver, and brain). HHT also may be associated with other diseases, such as juvenile polyposis syndrome and primary pulmonary hypertension.7
EPISTAXIS
Epistaxis occurs secondary to telangiectasias that develop on the nasal mucosa (Figure 1). Air passing through the nose leads to dryness and rupture of these fragile blood vessels. Epistaxis is the most common symptom among patients with HHT, occurring in 90 percent of patients,8 but it has a wide range of severity, even among affected family members. In some patients, it may be little more than a social nuisance; in others, it can be life threatening. Epistaxis tends to worsen with age, but 10 percent of patients with HHT will not have nosebleeds. These patients may mistakenly believe they do not have the disease, which could allow other manifestations of the disease to go undiagnosed.
Figure 1.
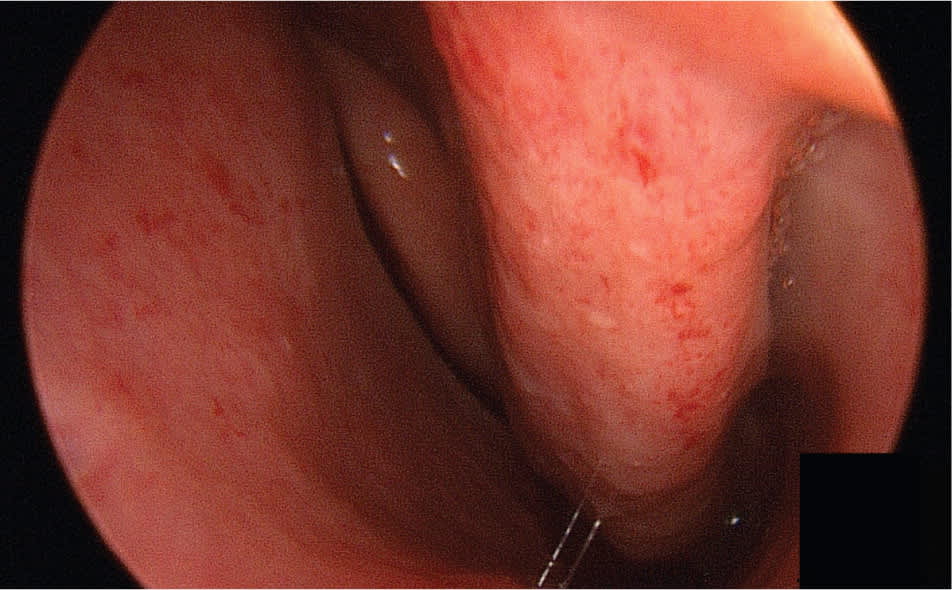
Multiple telangiectasias on the nasal mucosa.
Frequent epistaxis is a common cause of iron deficiency anemia in patients with HHT. Although epistaxis is common and most persons affected do not have HHT, the diagnosis should be suspected when there is a history of recurrent and unprovoked nosebleeds, skin or mucosal telangiectasias, a family history of epistaxis, or when epistaxis occurs in association with a personal or family history of other organ dysfunction consistent with HHT (e.g., sudden death, stroke, hypoxemia, liver disease).
TELANGIECTASIAS
Telangiectasias can occur anywhere on the skin but are most common on the face, chest, and hands. They generally develop by age 30 years and increase in number with age. Telangiectasias are bright red to purple and blanch with pressure. They can be visualized by transilluminating the patient's fingers with a small bright flash-light.9 Mucosal telangiectasias can occur anywhere along the GI tract and are often visible on the lips and tongue (Figure 2). GI bleeding occurs in approximately one third of patients with HHT and is another common cause of iron deficiency anemia.10
Figure 2.
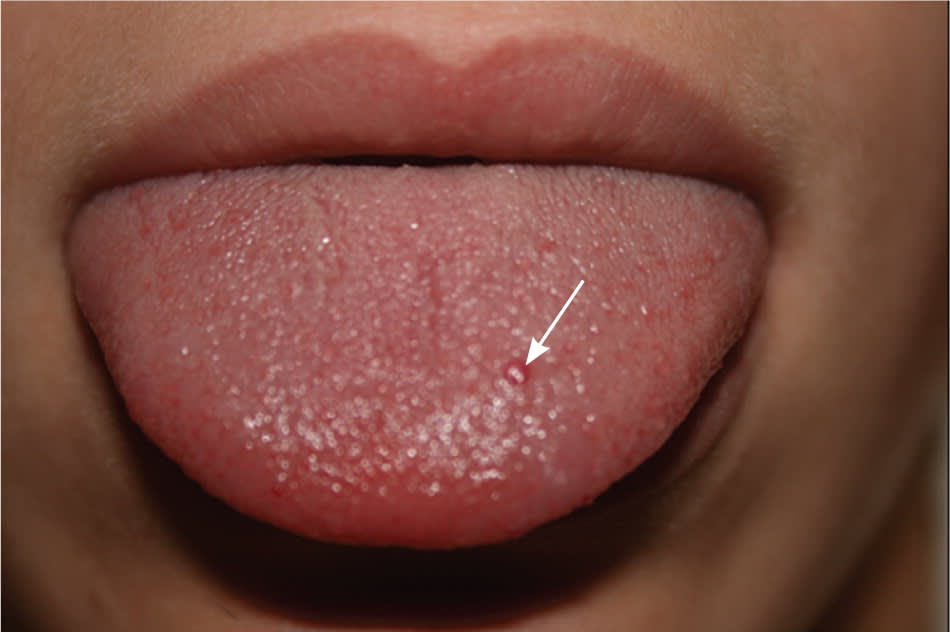
Single telangiectasia (arrow) involving the tongue of a 16-year-old girl.
PULMONARY ARTERIOVENOUS MALFORMATIONS
Pulmonary AVMs affect 15 to 30 percent of patients with HHT11 (Figure 3). The disorder should be considered in any patient diagnosed with a pulmonary AVM because 70 percent of these occur in patients with HHT.11 Pulmonary AVMs provide a right-to-left shunt between the pulmonary and systemic circulation, which can lead to hypoxemia. The normal pulmonary capillary bed prevents bacteria and emboli from reaching the systemic circulation. In the presence of a pulmonary AVM, this filtering function is absent. Patients with marked pulmonary AVMs can develop brain abscesses or experience cerebrovascular accidents. Cerebrovascular accidents may occur when materials that are normally filtered by the pulmonary vasculature (e.g., blood clot, bacteria) are allowed to pass through the pulmonary AVM to the left side of the heart and then to the brain. Patients with HHT should be screened for pulmonary AVMs.12–14 Contrast echocardiography (bubble echocardiography) is the most sensitive test to detect the presence of pulmonary AVMs. Saline (agitated with a small amount of air) is injected into the venous circulation during echocardiography. The presence of air bubbles in the left side of the heart after several cardiac cycles is consistent with the development of a pulmonary AVM. Because contrast echocardiography detects only the presence of pulmonary AVMs, a positive test should be followed by computed tomography angiography to determine the location and extent of the AVMs. It is important to diagnose and treat pulmonary AVMs in pregnant women because of the risk of hemorrhage during labor and delivery.15,16
Figure 3.
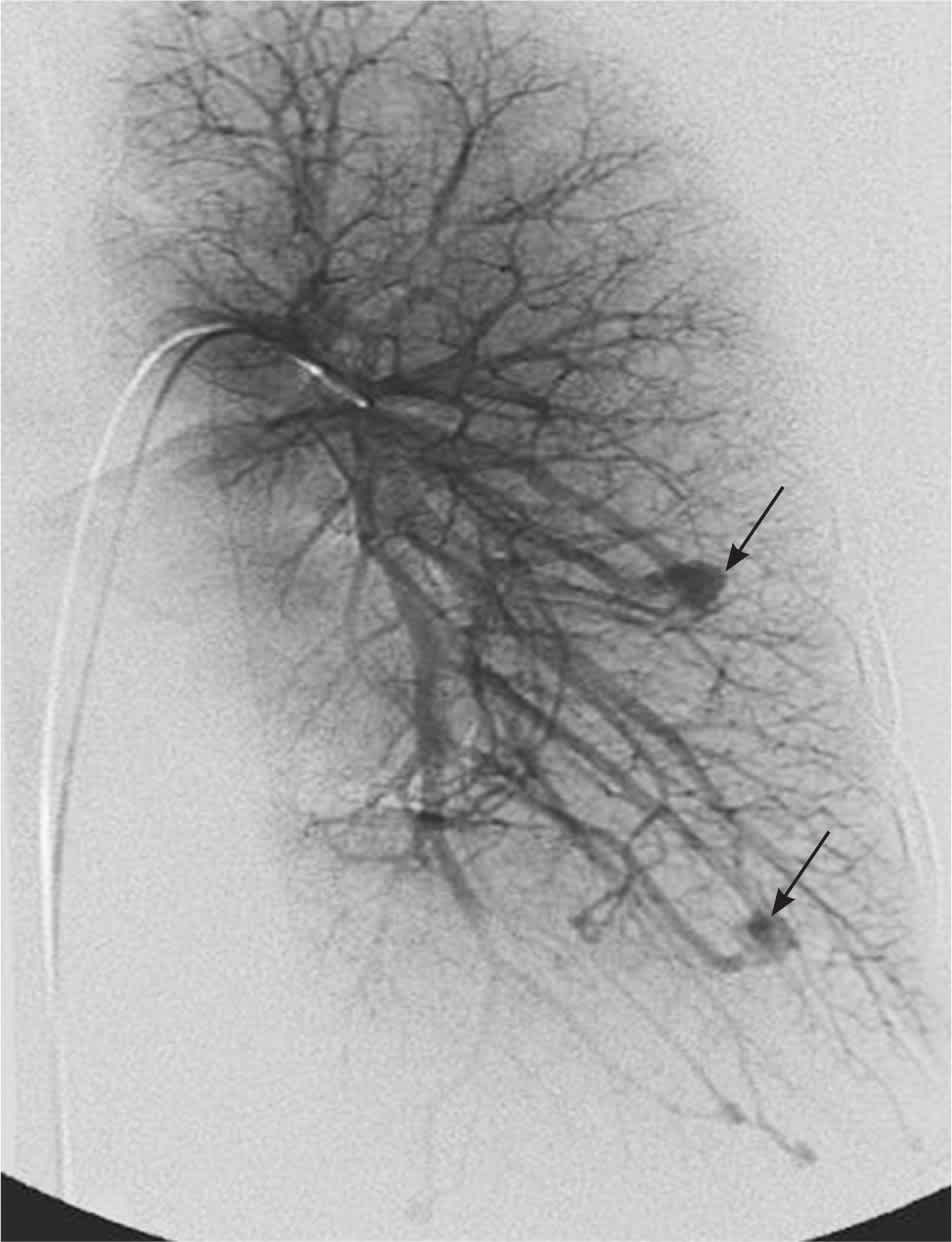
Angiogram demonstrating pulmonary arteriovenous malformations (arrows).
HEPATIC ARTERIOVENOUS MALFORMATIONS
Development of a hepatic AVM is estimated to occur in at least 30 percent of patients with HHT.17 AVMs and focal nodular hyperplasia in the liver may be detected during evaluation of an abnormal liver enzyme profile or as an incidental finding during an unrelated abdominal imaging study (Figure 4). Hepatic AVMs can lead to portal hypertension, biliary disease, and high output cardiac failure secondary to shunting between the hepatic artery and vein.18 A hepatic bruit may be present. Magnetic resonance imaging (MRI), computed tomography or Doppler ultrasonography may be used to document the presence of an AVM. Although these imaging studies may be grossly abnormal, only 5 percent of patients with hepatic involvement are symptomatic.19,20 Liver biopsy should not be performed in patients with suspected AVMs because of the risk of potentially fatal bleeding.21
Figure 4.
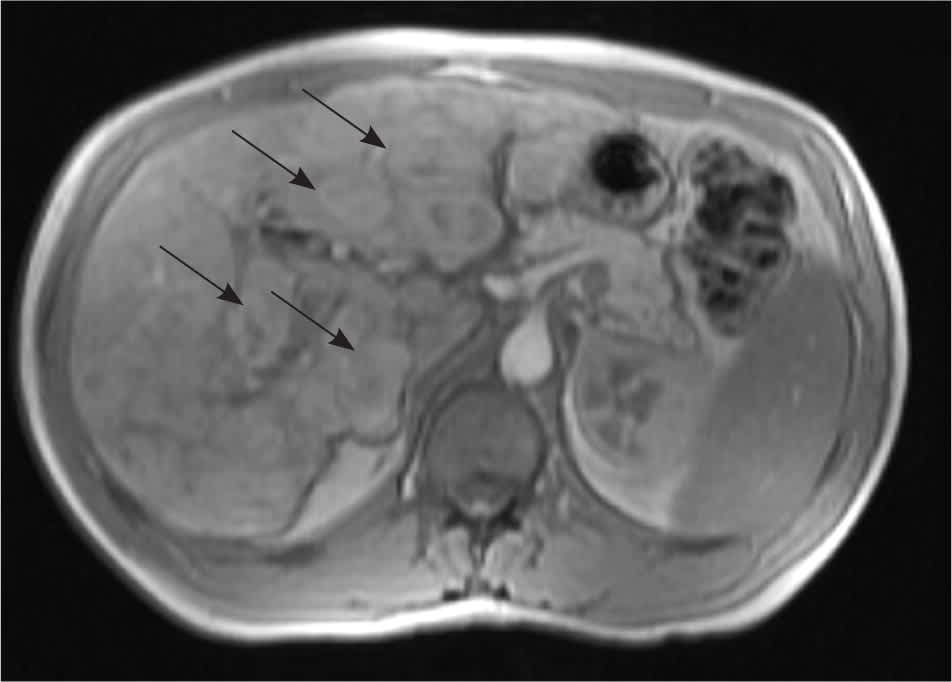
Magnetic resonance imaging of the liver showing multiple hepatic nodules (arrows) in an asymptomatic patient.
CEREBRAL ARTERIOVENOUS MALFORMATIONS
Cerebral AVMs affect more than 10 percent of patients with HHT.22 They can cause headache, seizures, and hemorrhage. Hemorrhage caused by rupture of cerebral AVMs can be devastating and lead to long-term disability or death. MRI is currently the best way to detect these vascular malformations.
Diagnosis and Screening
The Curaçao criteria were developed in 1999 for the diagnosis of HHT (Table 1).23 Global consensus guidelines for screening have been developed by HHT experts and follow evidence-based data when available.24 Genetic testing can be performed, but not every genetic mutation for HHT has been identified; therefore, some cases of HHT cannot be detected with current laboratory testing. If a mutation is found in one affected person, then other family members can be tested to determine if they carry the same mutation. The absence of the known mutation eliminates the need to screen for other manifestations of the disease. This can be especially helpful for children of affected parents because performing some of these tests may be more difficult and may require sedation or general anesthesia. Children of a parent with HHT who do not meet the criteria for a diagnosis of HHT should be considered to have the disease, unless excluded by genetic testing, for purposes of screening.25
Table 1. Diagnostic Criteria for HHT (Curaçao Criteria)

| Criteria | Characteristics |
|---|---|
| Epistaxis | Spontaneous, recurrent nosebleeds |
| Family history | A first-degree relative with HHT |
| Telangiectasias | Multiple telangiectasias at characteristic sites (e.g., lips, oral cavity, fingers, nose) |
| Visceral lesions | Examples include gastrointestinal telangiectasia (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, spinal AVM |
note: The HHT diagnosis is classified as definite if three criteria are present; possible or suspected if two criteria are present; and unlikely if fewer than two criteria are present.23
AVM = arteriovenous malformation; HHT = hereditary hemorrhagic telangiectasia.
Adapted with permission from Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91(1):67.
Screening should include contrast echocardiography and MRI of the brain.24 Transthoracic contrast echocardiography should be used as the initial screening test for pulmonary AVMs in patients with HHT.12,13,26 In young children, a determination of oxygen saturation on room air via pulse oximetry may be used until they are old enough to have contrast echocardiography. Patients with a negative result on contrast echocardiography should repeat the test every three to five years to monitor for the development of new pulmonary AVMs. Adults with possible or confirmed HHT should receive an MRI to screen for cerebrovascular AVMs using a protocol with and without contrast media administration and using sequences that detect blood products to maximize sensitivity. Children with possible or confirmed HHT should be screened for cerebrovascular malformations in the first six months of life (or at the time of diagnosis) with an unenhanced MRI. All patients with an MRI result positive for these lesions should be referred to a center with neurovascular expertise for consideration of invasive testing and further treatment.25–27 Cerebral AVMs are thought to be congenital and a negative MRI may not need to be repeated, although some recommend testing once during childhood and again in adulthood.
Screening for GI and liver involvement is recommended only for patients who demonstrate possible symptoms such as GI bleeding, anemia of unknown etiology, or hepatic or cardiac failure.
Treatment
Many patients with HHT have iron deficiency anemia caused by frequent epistaxis and GI bleeding. Treatment options include oral iron supplementation and intravenous iron infusion. Some patients may require blood transfusions. Because of the risk of anemia in patients with HHT, all patients older than 35 years should have annual measurement of hemoglobin or hematocrit levels.10
Treatment of epistaxis involves local measures, including the use of ointments to decrease drying of the nasal mucosa. Humidification of indoor air and avoidance of dry climates can also be beneficial. Because the nasal mucosa is a hormone-modulated tissue, estrogen may be used (topically or systemically) to help support the nasal mucosa and decrease bleeding. Laser coagulation of nasal telangiectasias may be helpful for patients who do not respond well to medical treatment and should be considered the first-line treatment option for HHT-related epistaxis requiring surgical intervention.28,29 Septal dermoplasty is a procedure in which the nasal mucosa is stripped from the nose and replaced with a thin dermis graft. The effects of laser coagulation and septal dermoplasty tend to be temporary and often require repeated treatment.29 The Youngs procedure involves complete closure of the nasal passages and can be curative30; however, patients become obligate mouth breathers and lose their sense of smell. Embolization may be useful as a short-term treatment for marked epistaxis, but results tend to be temporary and it should not be considered a first-line treatment.31
Pulmonary AVMs can be successfully embolized with coils.32 This is not a permanent fix and requires continued surveillance. The current recommendation is to treat any AVM that is at least 3 mm in diameter, although some physicians will treat AVMs as small as 1 mm. Patients who have AVMs, as well as those who have not yet been screened, should receive antibiotic prophylaxis before dental work or other “dirty” medical procedures to avoid the possible development of brain infection and abscess formation.24,33 Patients should have air filters on all intravenous lines, and it is recommended that they not scuba dive because of the risk of decompression sickness.34
GI bleeding may be treated locally with laser during endoscopy. Some lesions may be in the small intestine, making localization and treatment problematic. Hepatic disease leading to liver failure requires liver transplantation. Heart failure can be treated medically but may require heart transplantation.
Cerebral AVMs can be treated with embolization, radiotherapy, or surgery.
Future Treatments
Medical therapies will continue to be a source of interest in treating patients with HHT. Vascular endothelial growth factor inhibitors have shown promise in the treatment of epistaxis and advanced liver disease.35,36 The use of thalidomide (Thalomid) for GI bleeding is under investigation.37 The HHT Foundation International has helped fund a number of centers of excellence that provide a multidisciplinary approach to treatment and research of this disease. Patients are encouraged to be seen in these centers when the need arises and to remain in contact with them about new studies and developing treatments.
