
Am Fam Physician. 2013;87(3):191-197
Related Close-ups: Confronting Guillain-Barré Syndrome
Author disclosure: No relevant financial affiliations to disclose.
Guillain-Barré syndrome consists of a group of neuropathic conditions characterized by progressive weakness and diminished or absent myotatic reflexes. The estimated annual incidence in the United States is 1.65 to 1.79 per 100,000 persons. Guillain-Barré syndrome is believed to result from an aberrant immune response that attacks nerve tissue. This response may be triggered by surgery, immunizations, or infections. The most common form of the disease, acute inflammatory demyelinating polyradiculoneuropathy, presents as progressive motor weakness, usually beginning in the legs and advancing proximally. Symptoms typically peak within four weeks, then plateau before resolving. More than one-half of patients experience severe pain, and about two-thirds have autonomic symptoms, such as cardiac arrhythmias, blood pressure instability, or urinary retention. Advancing symptoms may compromise respiration and vital functions. Diagnosis is based on clinical features, cerebrospinal fluid testing, and nerve conduction studies. Cerebrospinal fluid testing shows increased protein levels but a normal white blood cell count. Nerve conduction studies show a slowing, or possible blockage, of conduction. Patients should be hospitalized for multidisciplinary supportive care and disease-modifying therapy. Supportive therapy includes controlling pain with nonsteroidal anti-inflammatory drugs, carbamazepine, or gabapentin; monitoring for respiratory and autonomic complications; and preventing venous thrombosis, skin breakdown, and deconditioning. Plasma exchange therapy has been shown to improve short-term and long-term outcomes, and intravenous immune globulin has been shown to hasten recovery in adults and children. Other therapies, including corticosteroids, have not demonstrated benefit. About 3 percent of patients with Guillain-Barré syndrome die. Neurologic problems persist in up to 20 percent of patients with the disease, and one-half of these patients are severely disabled.
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Diagnostic criteria for GBS include progressive, relatively symmetrical weakness with decreased or absent myotatic reflexes; symptoms must reach maximal intensity within four weeks of onset and other possible causes must be excluded. | C | 1, 2 |
| Lumbar puncture should be performed in all patients with suspected GBS. Cerebrospinal fluid protein levels may be normal in early GBS, but they are elevated in 90 percent of patients by the end of the second week of symptoms. | C | 7 |
| Supportive care of patients hospitalized with acute GBS should include anticoagulation and graduated compression stockings to prevent venous thrombosis. These patients also should be monitored for autonomic disturbances, including changes in blood pressure and pulse rate (especially bradycardia) and respiratory, bowel, and bladder dysfunction. | C | 23 |
| Plasma exchange is first-line therapy for GBS and should begin within seven days of symptom onset. | A | 30–33 |
| Intravenous immune globulin therapy is recommended for patients with GBS who require assistance with walking within two weeks of symptom onset. | C | 31 |
Definitions
The diagnostic criteria for GBS include progressive, relatively symmetrical weakness with decreased or absent myotatic reflexes. For diagnosis, symptoms must reach maximal intensity within four weeks of onset and other possible causes must be excluded.1,2 Table 1 includes features necessary for the diagnosis, and features that make the diagnosis unlikely or exclude it.2–9 This syndrome has several distinct subtypes (Table 2).4,5 Acute inflammatory demyelinating polyradiculoneuropathy (AIDP) is the most common subtype in the United States.3

| Feature | Comments | |
|---|---|---|
| Required for diagnosis | ||
| Bilateral symptoms | Usually begins in the legs | |
| Decreased myotatic reflexes | Complete areflexia often occurs in affected limbs | |
| Subacute, weakness and diminished/absent reflexes | Peaks within four weeks: peaks by two weeks in 50 percent of cases, and by three weeks in 80 percent of cases2–4 | |
| Supportive of diagnosis | ||
| Clinical | ||
| Autonomic involvement | Cardiac arrhythmias, orthostasis, blood pressure instability, urinary retention, slowing of gastrointestinal motility; absent in some subtypes | |
| Cranial nerve involvement | Facial weakness occurs in 30 to 50 percent of cases5,6; ophthalmoplegia is common with the Miller Fisher subtype; rarely an initial feature | |
| Relatively symmetrical | Symptoms may not be absolutely symmetrical in affected limbs or face | |
| Sensory involvement | Usually mild; absent in some subtypes, prominent in others (i.e., acute motor-sensory axonal neuropathy) | |
| Symptom pattern over time | Peak by two to four weeks, with variable plateau followed by recovery; permanent sequelae are possible | |
| Cerebrospinal fluid findings | ||
| Elevated protein levels | Levels may be normal early, but they are elevated by the end of the second week of symptoms in 90 percent of cases7 | |
| Normal white blood cell count | Less than 10 per mm3 (10 × 106 per L) | |
| Slowing (< 60 percent normal velocity) or blockage of nerve conduction | Slowing of nerve conduction occurs in 80 percent of cases, but this may take weeks to develop8,9 | |
| Rules out diagnosis | ||
| Evidence of neurotoxic conditions | Porphyria, diphtheria, botulism, toxic neuropathy | |
| Hexacarbon or lead exposure | Solvent abuse, exposure to some paints and vapors, occupational exposure | |
| Only sensory symptoms | Sensory symptoms are predominant in the very rare form of acute motor-sensory axonal neuropathy, but other neurologic symptoms are also present | |
| Raises doubt about diagnosis | ||
| Cerebrospinal fluid leukocytosis | Polymorphonuclear leukocyte or monocytes | |
| Delineated level of sensory dysfunction | — | |
| Persistent and significant asymmetry of symptoms | — | |
| Prominent bowel or bladder symptoms | Occurring at onset or persistent symptoms | |
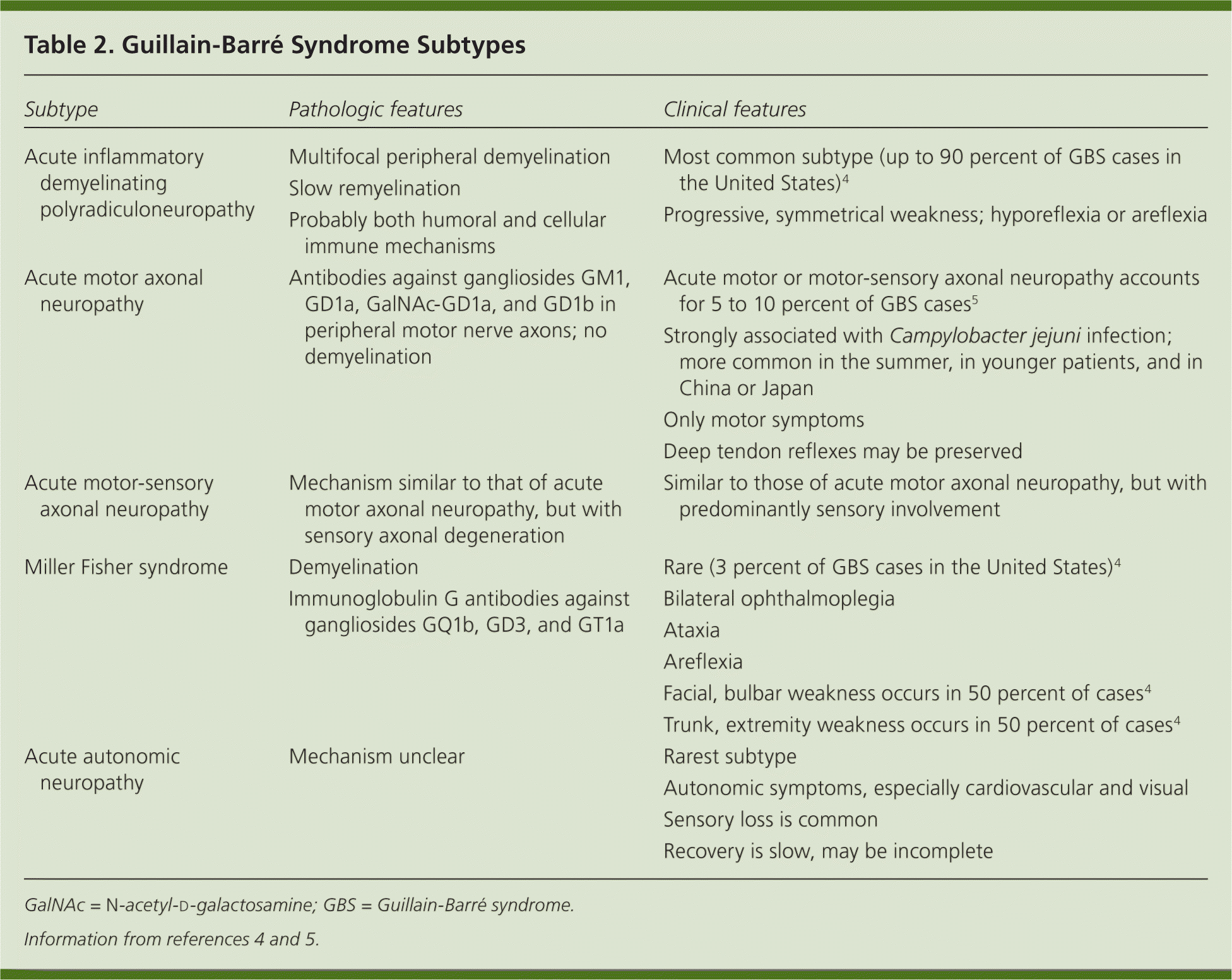
| Subtype | Pathologic features | Clinical features |
|---|---|---|
| Acute inflammatory demyelinating polyradiculoneuropathy |
|
|
| Acute motor axonal neuropathy |
|
|
| Acute motor-sensory axonal neuropathy |
|
|
| Miller Fisher syndrome |
| |
| Acute autonomic neuropathy |
|
|
Epidemiology
Several infections have been implicated in the development of GBS. About two-thirds of patients with the disease report respiratory or gastrointestinal symptoms in the three weeks before the onset of GBS symptoms.4 The strongest evidence implicates Campylobacter jejuni infection, but GBS also has been reported following infection with Mycoplasma pneumoniae, Haemophilus influenzae, cytomegalovirus, and Epstein-Barr virus.11 The reported associations of GBS with age, location, and season probably reflect the epidemiology of the precipitating conditions. Stressful events and surgeries also have been shown to trigger the disease.
Despite case reports of GBS developing after immunization for tetanus, hepatitis, and influenza, studies show that these immunizations lead to no or very little increased risk.12,13 The most recent study estimated that the attributable risk with 2009 pandemic influenza A (H1N1) immunization is up to two cases per 1 million doses, mainly in older persons.14
Etiology and Pathophysiology
The mechanism of GBS is believed to be an inflammatory neuropathy due to cross reactivity between neural antigens and antibodies that is induced by specific infections. Infectious organisms, such as C. jejuni, express lipooligosaccharides in the bacterial wall similar to gangliosides. This molecular mimicry creates antiganglioside antibodies that attack nerves. The specific antibody that is stimulated and its target area in the nerve may explain the different subtypes of GBS. Fewer than one per 1,000 patients with C. jejuni infection develops GBS, suggesting that host factors play a significant role in the pathologic process. However, research has not yet identified factors that increase an individual's risk of developing GBS.
GBS has been shown to cause symptoms through multifocal areas of mononuclear cell infiltration in the peripheral nerves. The location and severity of the inflammation correspond to the clinical manifestations. In AIDP, the myelin is predominantly damaged, whereas in acute motor axonal neuropathy, the nodes of Ranvier are targeted.5,15
Clinical Features
PRECEDING OR TRIGGERING CONDITIONS
The most common symptoms reported before onset of GBS are fever, cough, sore throat, and other upper respiratory symptoms.16 Infection with Epstein-Barr virus has been linked to milder forms of GBS.17 Gastrointestinal symptoms may be more likely to precede the acute motor or motor-sensory axonal neuropathy subtypes that are related to slower recovery and higher risk of residual disability.18 A timely diagnosis of GBS depends on a high index of suspicion for the condition.
CLINICAL PRESENTATION
Initial symptoms typically include weakness, numbness, tingling, and pain in the limbs. The extent, progression, and severity of symptoms vary greatly among individual patients. The most prominent symptom in patients with AIDP is bilateral, relatively symmetrical weakness of the limbs that progresses rapidly. In up to 90 percent of patients with AIDP, symptoms begin in the legs and advance proximally. The advancing weakness may compromise respiratory muscles, and about 25 percent of patients who are hospitalized require mechanical ventilation.4 Respiratory failure is reported to be more common in patients with rapid progression of symptoms, upper limb weakness, autonomic dysfunction, or bulbar palsy. The weakness typically reaches its peak by the second week, followed by a plateau of variable duration before resolution or stabilization with residual disability.19
Facial, oropharyngeal, and oculomotor muscles may be affected because of cranial neuropathy, especially in the less common subtypes. Paresthesia in the feet and hands is common, but sensory symptoms are generally mild, except for in those patients with the acute motor-sensory axonal neuropathy subtype. Autonomic symptoms occur in about two-thirds of patients and include cardiac arrhythmias, orthostasis, blood pressure instability, urinary retention, and slowing of gastrointestinal motility.20
Pain, especially with movement, is reported by 50 to 89 percent of patients with GBS. The pain is described as severe, deep, aching, or cramping (similar to sciatica) in the affected muscles or back, and is often worse at night. Because the pain is nociceptive and/or neuropathic, it may be difficult to control.6 Pain as an initial or prominent early symptom may delay diagnosis of GBS.
Diagnostic Investigations
Diagnosis of GBS depends on repeated neurologic examinations demonstrating a classic pattern of advancing, symmetrical motor weakness and diminished myotatic reflexes. Specific changes in cerebrospinal fluid (CSF) measurements and nerve conduction studies are strongly supportive of the diagnosis (Table 12–9). Lumbar puncture and neurophysiology testing should be performed in all patients with suspected GBS.7 Additional testing can be used on an individual basis to rule out alternative explanations for GBS-like symptoms. Table 3 presents the differential diagnosis of the disease.
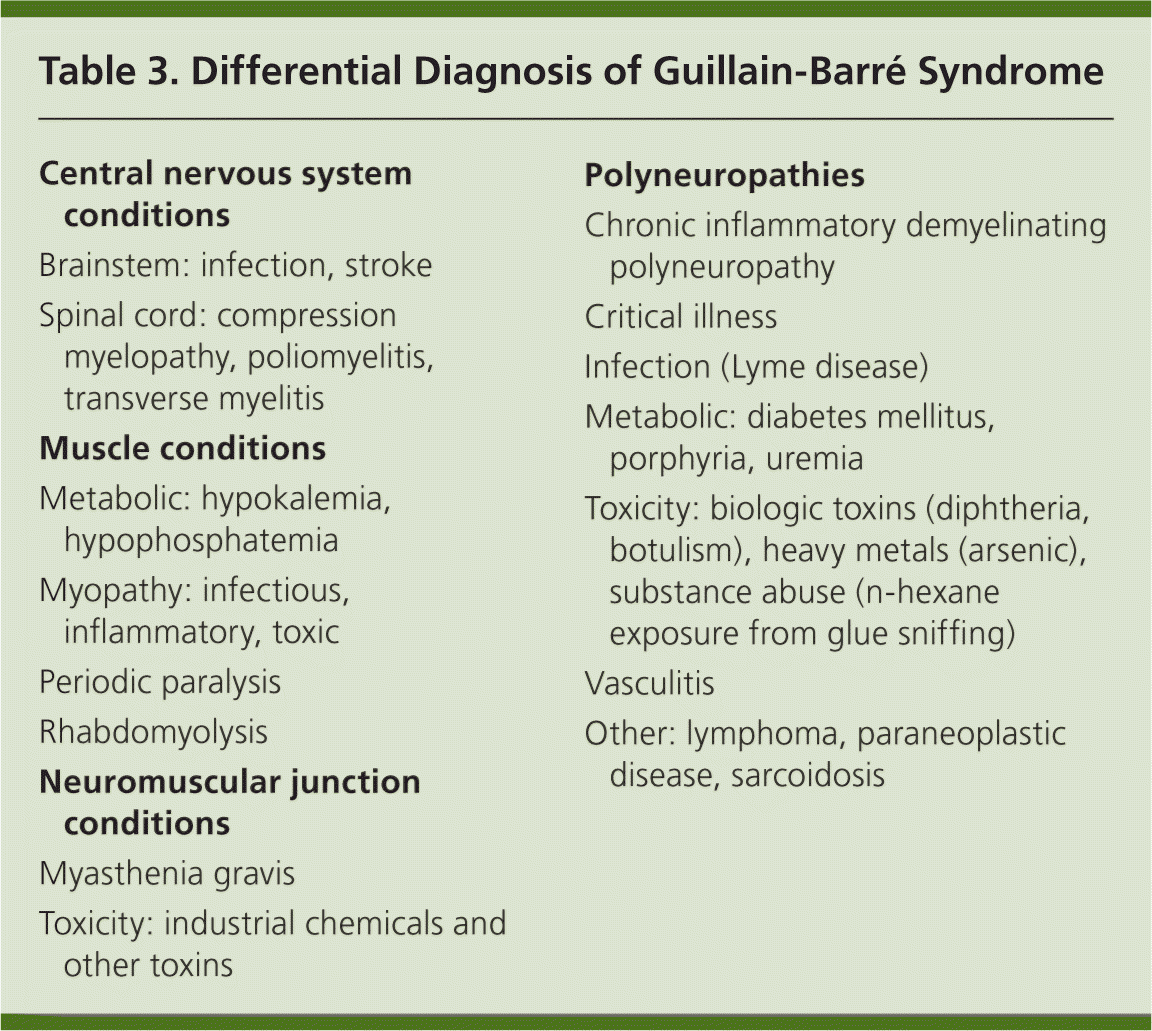
| Central nervous system conditions | |
| Brainstem: infection, stroke | |
| Spinal cord: compression myelopathy, poliomyelitis, transverse myelitis | |
| Muscle conditions | |
| Metabolic: hypokalemia, hypophosphatemia | |
| Myopathy: infectious, inflammatory, toxic | |
| Periodic paralysis | |
| Rhabdomyolysis | |
| Neuromuscular junction conditions | |
| Myasthenia gravis | |
| Toxicity: industrial chemicals and other toxins | |
| Polyneuropathies | |
| Chronic inflammatory demyelinating polyneuropathy | |
| Critical illness | |
| Infection (Lyme disease) | |
| Metabolic: diabetes mellitus, porphyria, uremia | |
| Toxicity: biologic toxins (diphtheria, botulism), heavy metals (arsenic), substance abuse (n-hexane exposure from glue sniffing) | |
| Vasculitis | |
| Other: lymphoma, paraneoplastic disease, sarcoidosis | |
Patients with GBS classically have increased protein levels and a normal white blood cell count (i.e., less than 10 per mm3 [10 × 106 per L]) in CSF. Protein levels in CSF may be normal in early GBS, but they are elevated in 90 percent of patients by the end of the second week of symptoms.7 The normal CSF white blood cell count helps differentiate GBS from other infectious, inflammatory, and malignant diseases. However, GBS may produce an elevated CSF white blood cell count in patients who are serologically positive for human immunodeficiency virus.21
Slowing of nerve conduction occurs in an estimated 80 percent of patients with GBS.2 Electrodiagnostic study results may be normal in up to 13 percent of patients soon after symptom onset, but rarely remain normal on sequential testing over the initial weeks of symptoms.8 The sequential study findings depend on the subtype and severity of GBS, but they most commonly show multifocal demyelinating polyneuropathy with secondary axonal degeneration followed by recovery. Repeated electrodiagnostic studies may help determine the GBS subtype and predict prognosis.8,9 To provide adequate data, at least three sensory and three motor nerves should be tested, although the sural nerve should be avoided because it often remains normal in GBS.19
Management
Most patients with GBS recover spontaneously. However, because of the unpredictable course and potential for death or significant disability, all patients with the disease should be hospitalized for multidisciplinary supportive care and disease-modifying therapy.22
SUPPORTIVE CARE
Consensus guidelines recommend subcutaneous anticoagulation and graduated compression stockings for patients who are hospitalized to decrease the risk of venous thrombosis.23 Patients hospitalized with GBS also should be monitored for autonomic disturbances, including changes in blood pressure and pulse rate (especially bradycardia) and respiratory, bowel, and bladder dysfunction.23 Respiratory compromise can develop rapidly and may be more likely in patients with rapid progression of symptoms, bulbar palsy, and upper limb and autonomic symptoms. Predictive factors that indicate the need for mechanical ventilation are shown in Table 4.19,24–26 Early tracheostomy should be considered in patients who may need prolonged ventilator support.23,27 Swallowing evaluation should be performed in patients with facial or oropharyngeal weakness because of the risk of aspiration. Patients with limited mobility should be closely monitored and prospectively treated to prevent skin breakdown.

| Predicts the need for mechanical ventilation24–26 |
| Bulbar symptoms |
| Inability to raise the head or flex the arms |
| Inadequate cough |
| Maximum expiratory pressure: < 40 cm H2O |
| Maximum inspiratory pressure: < 30 cm H2O |
| Time from onset of symptoms to hospital admission is less than seven days |
| Vital capacity: < 60 percent of predicted or < 20 mL per kg |
| Vital capacity, maximum inspiratory pressure, or maximum expiratory pressure reduced by at least 30 percent |
| Predicts long-term disability19,24 |
| Absence of motor response |
| Antecedent diarrheal illness |
| Axonal involvement |
| Campylobacter jejuni or cytomegalovirus infection |
| Inability to walk at 14 days |
| Older age |
| Rapid progression of symptoms |
| Severity of symptoms at their peak |
Patients with GBS often experience both neuropathic and nociceptive pain, and one-half of patients rate the pain as severe.23 Nonsteroidal anti-inflammatory drugs or opioid medications may not provide adequate relief, and opioids may exacerbate autonomic symptoms, especially constipation. Small studies reported that, when added to other analgesic regimens, gabapentin (Neurontin; 15 mg per kg daily)28 or carbamazepine (Tegretol; 300 mg daily for three days)29 was beneficial in patients with acute GBS in intensive care units. Long-term management with tricyclic antidepressants, tramadol (Ultram), gabapentin, or carbamazepine may be beneficial for chronic pain.
Patients with GBS should do strengthening exercises during the acute phase, and rehabilitation should be considered to regain mobility and function as they improve. Up to 80 percent of patients experience persistent, severe fatigue after resolution of other symptoms. The degree of fatigue does not appear to be related to severity of illness, duration of disability, or patient age. No pharmacologic treatments have proved beneficial for fatigue. Despite limited evidence, a supervised exercise program is recommended to improve fatigue and functional abilities.26
DISEASE-MODIFYING THERAPY
Specific treatments to hasten recovery and/or ameliorate symptoms target the aberrant immune response in GBS. Removing circulating immune complexes via plasma exchange has been shown to improve the time to recover the ability to walk, the need for artificial ventilation, the duration of ventilation, and measured muscle strength after one year compared with placebo.30,31 According to new evidence-based guidelines, plasma exchange is effective and should be used for severe AIDP, and is probably effective and should be considered for mild AIDP.32 Optimal response is achieved when plasma exchange is performed within seven days of symptom onset, although there is benefit up to 30 days after symptom onset.30–33 Some evidence suggests that patients with mild GBS benefit from two sessions of plasma exchange, whereas patients with moderate to severe disease appear to require four sessions.33 The role of plasma exchange in children has not been established.
Intravenous immune globulin therapy has been shown to hasten recovery in adults and children compared with supportive therapy alone. The typical dosage is 400 mg per kg per day for five days, although some evidence suggests that a total of 2 g per kg over two days is equally effective.34,35 Intravenous immune globulin therapy is easier to manage than plasma exchange and has significantly fewer complications.31 The initial response does not necessarily predict the outcome because patients may stabilize or continue to decline after the therapy. Intravenous immune globulin therapy should be started within two weeks of symptom onset, and should be considered for patients who are nonambulatory. The therapy may have a role two to four weeks after symptom onset as well, but the evidence of effectiveness is weaker.31
The few studies of plasma exchange followed by intravenous immune globulin therapy have not shown benefit over monotherapy. Therefore, sequential therapy is not recommended.31
Corticosteroids are not recommended for the treatment of GBS.31,35 A systematic review of six clinical trials that included 587 patients treated with different dosages and forms of corticosteroids reported no difference in mortality or disability outcome between patients taking corticosteroids and those taking placebo.35 Four additional studies demonstrated that patients receiving corticosteroids had less improvement in disability after four weeks of therapy than patients not treated with corticosteroids, leading to concerns that they may delay long-term recovery.3 Other therapies have been studied, but the evidence is limited and of low quality.36,37
Prognosis
Even with treatment, about 3 percent of patients with GBS die. The median hospital stay is seven days, and up to 25 percent of patients require intubation and mechanical ventilation. The prognosis is worse in older patients, those with severe symptoms, and those with rapid onset of symptoms. Neurologic problems persist in up to 20 percent of patients, one-half of whom are severely disabled.3,10 Predictive factors that indicate a higher risk of long-term disability are shown in Table 4.19,24–26 Resources for patients with GBS are available from the National Institute of Neurological Disorders and Stroke at http://www.ninds.nih.gov/disorders/gbs/gbs.htm, and from GBS/Chronic Inflammatory Demyelinating Polyneuropathy Foundation International at http://www.gbs-cidp.org.
Data Sources: A PubMed search was completed in Clinical Queries using the key terms Guillain Barré, diagnosis, and treatment. The search included meta-analyses, randomized controlled trials, clinical trials, and reviews. We also searched the Agency for Healthcare Research and Quality evidence reports, Clinical Evidence, the Cochrane database, Essential Evidence Plus, the National Guideline Clearinghouse database, and the bibliographies of the initially identified papers. Search date: November 15, 2011.
