
Am Fam Physician. 2013;88(5):319-327
Patient information: See related handout on prolactinoma, written by the authors of this article.
Author disclosure: No relevant financial affiliations.
Prolactinomas and nonfunctioning adenomas are the most common types of pituitary adenomas. Patients with pituitary adenomas may present initially with symptoms of endocrine dysfunction such as infertility, decreased libido, and galactorrhea, or with neurologic symptoms such as headache and visual changes. The diagnosis may also be made following imaging done for an unrelated issue in an asymptomatic patient; this is termed a pituitary incidentaloma. Oversecretion of hormones from a dysfunctional pituitary gland may result in classic clinical syndromes, the most common of which are hyperprolactinemia (from oversecretion of prolactin), acromegaly (from excess growth hormone), and Cushing disease (from overproduction of adrenocorticotropic hormone). In the diagnostic approach to a suspected pituitary adenoma, it is important to evaluate complete pituitary function, because hypopituitarism is common. Therapy for pituitary adenomas depends on the specific type of tumor, and should be managed with a team approach to include endocrinology and neurosurgery when indicated. Dopamine agonists are the primary treatment for prolactinomas. Small nonfunctioning adenomas and prolactinomas in asymptomatic patients do not require immediate intervention and can be observed.
Pituitary adenomas are the most common type of pituitary disorder.1 They are benign neoplasms that account for 10% to 15% of all intracranial masses. Few large studies have delineated the exact prevalence, but a recent study of the inhabitants of a community in the United Kingdom found the overall prevalence to be higher than previously reported, at 77.6 per 100,000 persons.2 Although autopsy and radiologic studies indicate that the prevalence may be as high as 20%, the majority of these tumors are incidentalomas without clinical significance.3,4 Whether during an evaluation of symptoms commonly presented to them, or through incidental discovery of a pituitary mass, family physicians are often the first to consider the diagnosis of a pituitary adenoma.
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Serum prolactin level should be measured in all patients with signs or symptoms of pituitary adenoma. | C | 7, 9, 34 |
| Evaluation of a suspected pituitary mass should include magnetic resonance imaging. | C | 7, 21 |
| Patients with pituitary adenomas affecting the optic chiasm on imaging should have formal visual field testing. | C | 7 |
| Symptomatic prolactinomas and macroprolactinomas should be treated medically with dopamine agonists. | B | 9, 17 |
| Cabergoline is the preferred dopamine agonist for the treatment of prolactinomas. | B | 23, 24 |
| Patients who have growth hormone– and adrenocorticotropic hormone–secreting tumors and those with symptomatic nonfunctioning macroadenomas should be referred for surgical removal. | B | 7, 25, 30 |
Classification
Pituitary adenomas are categorized based on primary cell origin and type of hormone secreted (Table 15–12 ). If the adenoma does not secrete a sufficient level of hormones to be detectable in the blood or to result in clinical manifestations, it is considered nonfunctioning. Prolactinomas comprise 40% to 57% of all adenomas, followed by nonfunctioning adenomas (28% to 37%), growth hormone–secreting adenomas (11% to 13%), and adrenocorticotropic hormone (ACTH)–secreting adenomas (1% to 2%). Pituitary adenomas that secrete follicle-stimulating hormone (FSH), luteinizing hormone (LH), or thyroid-stimulating hormone (TSH) are rare.2,13 Tumors are also categorized based on size. If the tumor is 10 mm or larger, it is considered a macroadenoma; if it is less than 10 mm, it is considered a microadenoma. Microadenomas are slightly more common than macroadenomas (57.4% vs. 42.6%).13
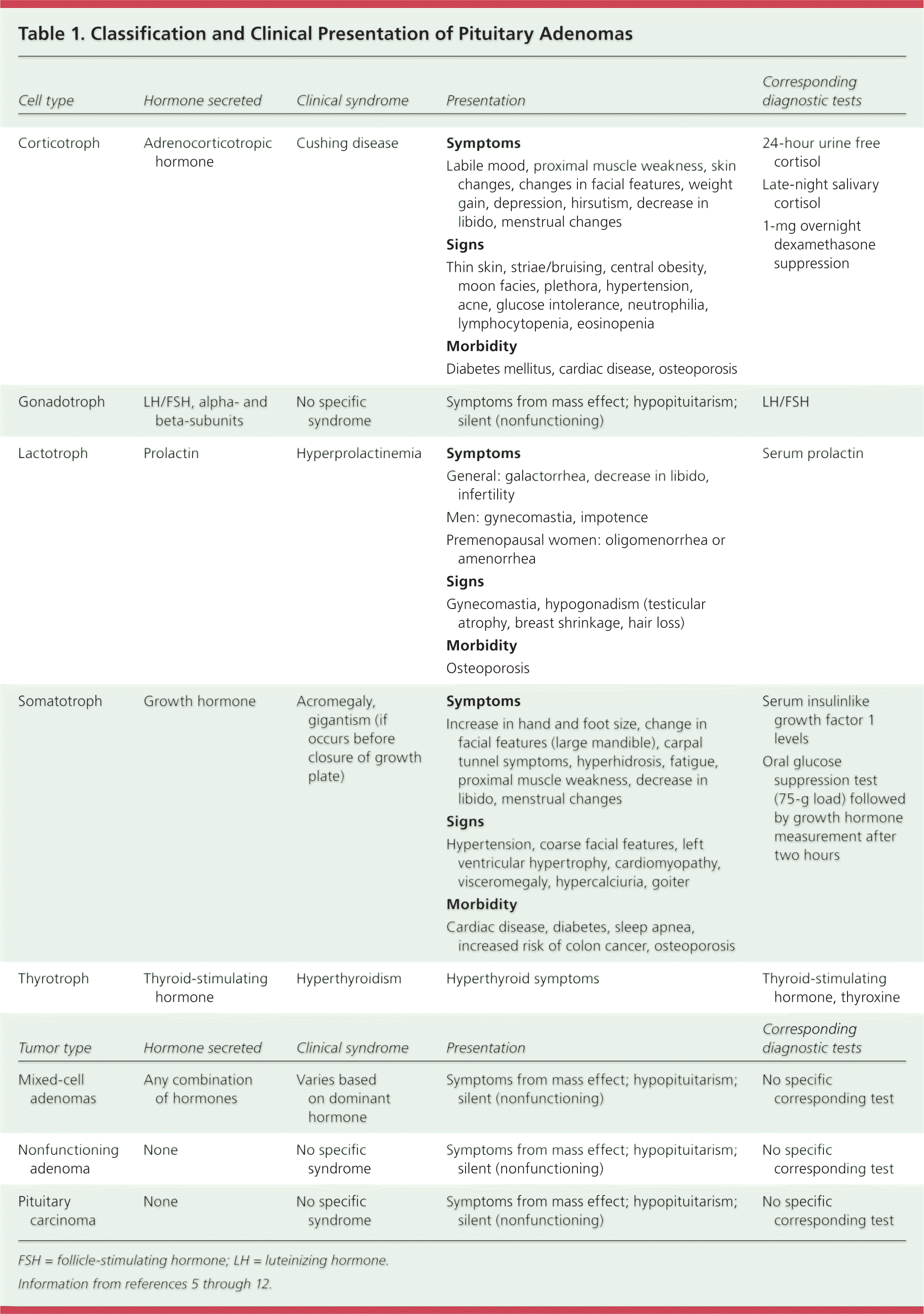
| Cell type | Hormone secreted | Clinical syndrome | Presentation | Corresponding diagnostic tests |
|---|---|---|---|---|
| Corticotroph | Adrenocorticotropic hormone | Cushing disease | Symptoms
|
|
| Gonadotroph | LH/FSH, alpha- and beta-subunits | No specific syndrome | Symptoms from mass effect; hypopituitarism; silent (nonfunctioning) |
|
| Lactotroph | Prolactin | Hyperprolactinemia | Symptoms
|
|
| Somatotroph | Growth hormone | Acromegaly, gigantism (if occurs before closure of growth plate) | Symptoms
|
|
| Thyrotroph | Thyroid-stimulating hormone | Hyperthyroidism | Hyperthyroid symptoms |
|
Pathophysiology
The pituitary gland sits inferior to the hypothalamus (Figure 1). It is surrounded caudally by the sphenoid bone in a basketlike structure called the sella turcica, and superiorly by the optic chiasm. The sella turcica forces an expanding adenoma superiorly, leading to compression of the optic nerve and headaches from mass effect. Additionally, destruction or compression of the pituitary gland may cause complete or partial hypopituitarism.5
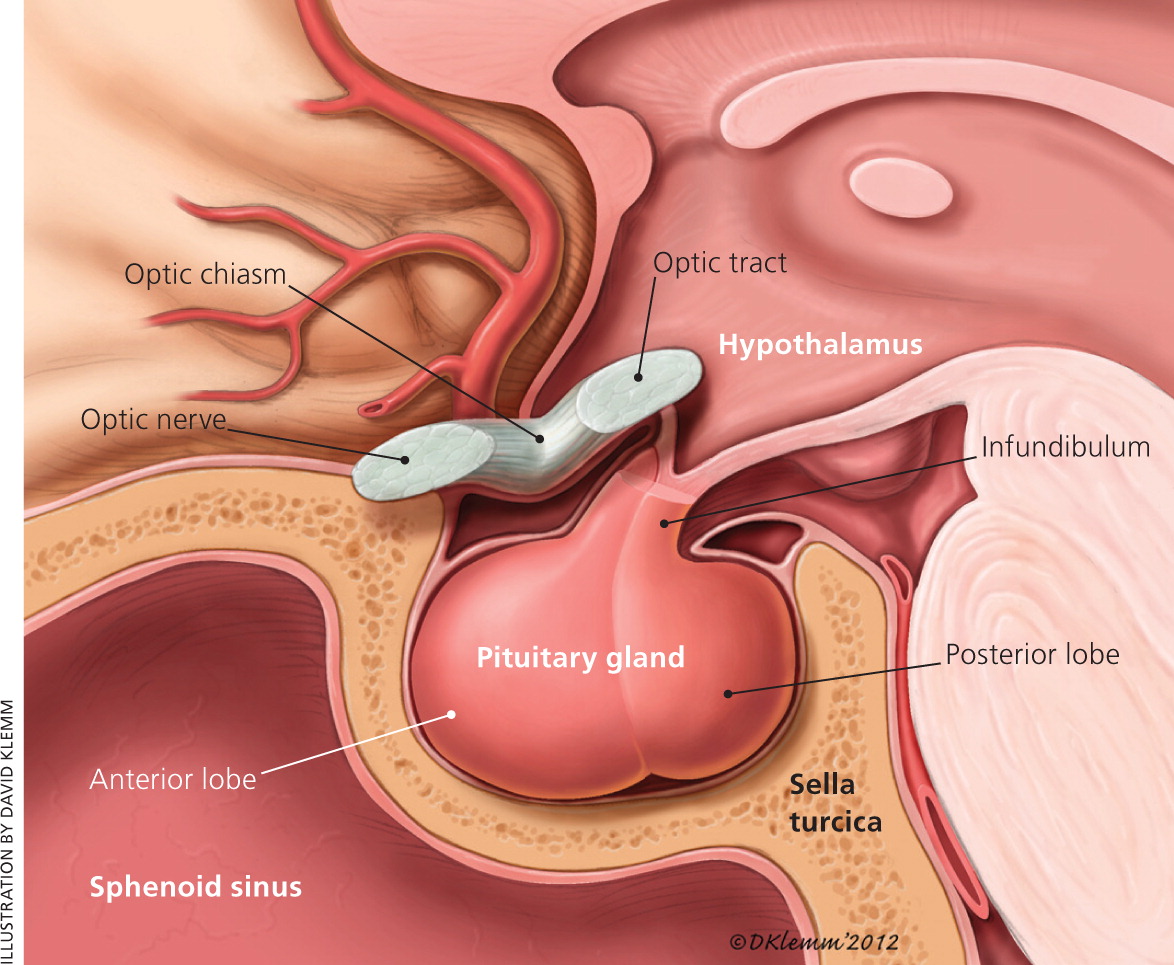
Pituitary adenomas are benign tumors that arise from one of the five cell types that comprise the anterior pituitary (lactotrophs, gonadotrophs, somatotrophs, corticotrophs, and thyrotrophs). Tumors rarely form from a combination of these cells. Pituitary adenomas are true neoplasms with a monoclonal cell origin.14 Hypersecretion or diminished inhibition of the hormones of the hypothalamic-pituitary axis can lead to the constellation of endocrine symptoms often seen in patients with pituitary adenomas.
Clinical Features
Pituitary adenomas present clinically in three ways: syndromes of hormone hypersecretion or deficiency; neurologic manifestations from mass effect of an expanding gland; or an incidental finding on imaging done for an unrelated issue.
HORMONAL
A pituitary adenoma may present as a distinct syndrome of hormone hypersecretion, the most common of which are hyperprolactinemia, acromegaly, and Cushing disease (Table 15–12 ). Another hormonal manifestation of a pituitary adenoma is either partial or complete hypopituitarism, most often hypogonadism. This is a result of interference with normal hormone secretion, either from direct compression of the pituitary gland or (in the case of hyperprolactinemia) inhibition of the pulsatile secretion of LH, leading to inadequate gonadal stimulation.6 The clinical presentation depends on the patient's sex. In women, the clinical presentation also depends on whether the patient is premenopausal or postmenopausal. Fatigue and loss of libido are common in men and women. Men may have erectile dysfunction, whereas premenopausal women often experience oligomenorrhea or amenorrhea6,11 (Table 15–12 ).
NEUROLOGIC
The most common neurologic symptoms in patients with pituitary adenomas are headaches and visual changes. Neurologic symptoms are more common in nonfunctioning adenomas or gonadotroph adenomas because these tumors do not secrete sufficient hormones to cause endocrine-type symptoms, and their diagnosis often is delayed until patients present with symptoms of mass effect.11 Headaches, which are believed to result from stretching of the dural sheath, are nonspecific and do not necessarily correlate with tumor size.5
When the tumor enlarges, it compresses the optic chiasm superiorly, primarily causing visual field deficits, most often bitemporal hemianopia.6 If the adenoma expands laterally into the cavernous sinus, it has the potential to affect the cranial nerves housed there, including cranial nerves III (oculomotor), IV (trochlear), and VI (abducens). With more severe compression or direct invasion of the optic nerve, decreased visual acuity may occur. In contrast with headaches, visual disturbances tend to correlate with tumor size. They also tend to occur insidiously, so that many patients are not aware of them until they are specifically tested.15 Other neurologic presentations, such as cerebrospinal fluid rhinorrhea, seizures, and pituitary apoplexy, can occur but are uncommon.
| Test | Indication | Abnormal (reference range)* | Clinical notes |
|---|---|---|---|
| 24-hour urine free cortisol | Cortisol excess | Elevated (10 to 84 mcg total per 24-hour period) | Urine collection must be accurate and total |
| High false-positive rate in women taking estrogen | |||
| Diagnostic if four times greater than normal | |||
| Adrenocorticotropic hormone | Determine source of excess cortisol | Elevated (6 to 48 pg per mL [1 to 11 pmol per L]) | Levels will be elevated with pituitary or ectopic source of excess cortisol |
| Estradiol | Hormone deficiency in females | Low (1.5 to 3 pg per mL [6 to 11 pmol per L])† | Not accurate in women taking oral contraceptives or hormone therapy |
| Values vary based on phase of menstrual cycle | |||
| Free T4 | Thyroid deficiency | Low (4.2 to 13 ng per dL [54 to 167 pmol per L]) | Low T4 with normal or low TSH indicates secondary hypothyroidism (possibly from pituitary dysfunction) |
| Late night salivary cortisol | Cortisol excess | Elevated (< 0.01 to 0.09 mcg per dL) | Midnight sample |
| Low-dose dexamethasone suppression | Initial test for cortisol excess | Elevated (serum cortisol ≥ 1.8 ng per dL) | 1 mg of dexamethasone given at 11 p.m., cortisol test at 8 a.m. |
| Abnormal if cortisol levels fail to decrease to < 1.8 ng per dL | |||
| High false-positive rate in women taking estrogen | |||
| Further testing needed to rule out the source of excess cortisol and to rule out “pseudo–Cushing syndrome” | |||
| Oral glucose suppression | Acromegaly (excess growth hormone) confirmatory test | Elevated (0 to 1 ng per mL)† | Failure of growth hormone to decrease to < 1 ng per mL two hours after administering 75 g of oral glucose |
| Serum alpha-subunit | Determine if nonfunctioning or gonadotroph-secreting tumor | Elevated (0.04 to 1.23 ng per mL)† | Test for the alpha subunit common to LH, FSH, TSH, and hCG |
| May be overproduced in some pituitary adenomas (most commonly gonadotroph- and thyrotroph-secreting types) | |||
| Useful if elevated LH/FSH levels and etiology of elevation is unknown | |||
| Serum cortisol | Cortisol deficiency | Low (8 to 19 mcg per dL [221 to 524 nmol per L]) | Early morning testing |
| If < 13 mcg per dL (359 nmol per L), patient should undergo dynamic testing | |||
| Serum insulinlike growth factor 1 | Excess growth hormone | Elevated (76 to 328 ng per mL [10 to 43 nmol per L])‡ | High sensitivity |
| Normally elevated during pregnancy | |||
| May be low in patients with poorly controlled diabetes mellitus, liver disease, hypothyroidism, and malnutrition | |||
| Serum LH/FSH | Gonadotroph-secreting tumor | Elevated | In postmenopausal women, elevated LH/FSH levels are normal |
| FSH (2 to 35 mIU per mL [2 to 35 IU per L])† | Value for menstruating women varies based on phase of menstrual cycle | ||
| LH (1.5 to 50 mIU per mL [1.5 to 50 IU per L])† | |||
| Hypogonadism | Low | ||
| FSH (2 to 35 mIU per mL)† | |||
| LH (1.5 to 50 mIU per mL)† | |||
| Serum macroprolactin | Hyperprolactinemia | Present | Biologically inactive form of prolactin, with no pathologic association |
| Useful to check in patients with hyperprolactinemia who have moderate elevations (100 to 300 mcg per L [4,348 to 13,043 pmol per L]) of serum prolactin with unknown etiology or when radiographic findings and clinical presentation are not consistent with serum prolactin level | |||
| Serum prolactin | Hyperprolactinemia | Elevated (3 to 24 mcg per L [130 to 1,043 pmol per L]) | ≥ 250 mcg per L (10,870 pmol per L) highly suggestive of prolactinoma |
| Moderate elevations (25 to 249 mcg per L [1,087 to 10,826 pmol per L]) should prompt investigation of other causes of hyperprolactinemia | |||
| Serum level correlates with tumor size; when level does not correlate with size, consider serial dilutions | |||
| Serum TSH | Evaluate thyrotroph-secreting tumor | Elevated (0.5 to 4.8 mIU per L) | May be atypically normal in relation to free T4 |
| Thyroid hormone deficiency | Low (0.5 to 4.8 mIU per L) | ||
| Testosterone (total) | Hormone deficiency in males | Low (350 to 1,030 ng per dL [12 to 36 nmol per L])‡ | Total (free + protein-bound) alone is generally an accurate picture of testosterone level |
| Testosterone (free) | Low (52 to 280 pg per mL)‡ | Measurements should be taken at 8 a.m. |
INCIDENTAL
Increased use and sensitivity of computed tomography (CT) and magnetic resonance imaging (MRI) have identified many pituitary lesions that otherwise might not have been detected. Autopsy and radiology studies suggest that between 10% and 20% of all pituitary adenomas may be unsuspected or found incidentally.3,4
Diagnostic Approach
HORMONAL SYMPTOMS
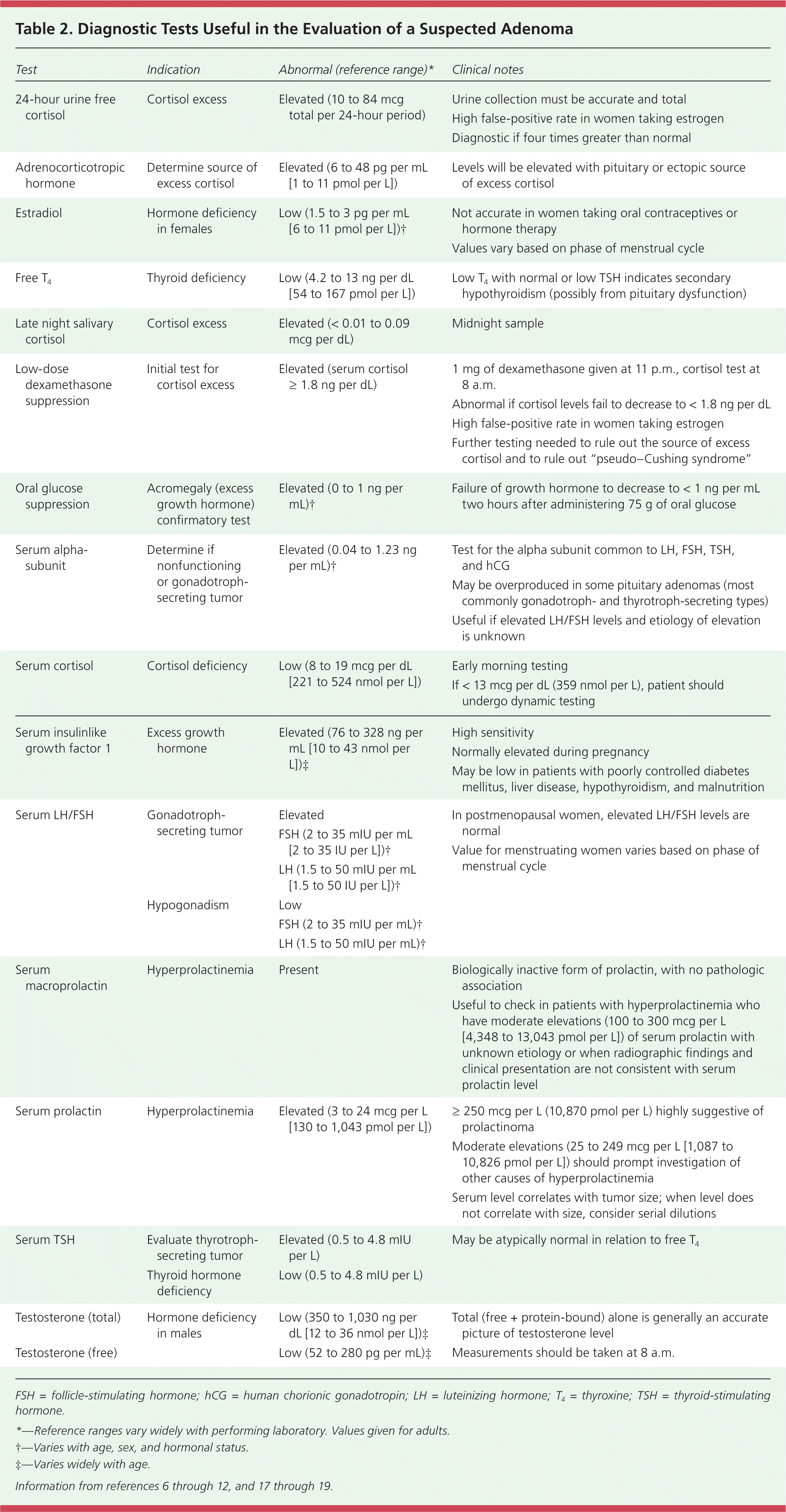
A patient who presents with symptoms of hormone excess likely has a functioning adenoma. Evaluation can be geared toward the specific hypersecretory syndrome (Table 15–12 ). Hormone deficiencies should also be evaluated because hypopituitarism is present in up to 30% of adenomas,13,16 and because of the need to address deficiencies in future treatment regimens. Consensus guidelines recommend obtaining an endocrine panel as an initial set of laboratory tests.7,9 This includes serum prolactin, insulinlike growth factor 1 (IGF-1), LH/FSH, TSH, thyroxine (T4), and an initial test for cortisol excess—a 24-hour urine free cortisol, a late night salivary cortisol, or a low-dose dexamethasone suppression test. The 24-hour urine free cortisol, late night salivary cortisol, and overnight dexamethasone suppression tests have similar accuracy (positive likelihood ratio [LR+] = 10.6, negative likelihood ratio [LR–] = – 0.16; LR+ = 8.8, LR– = 0.07; and LR+ = 16.4, LR– = 0.06, respectively).12 Dexamethasone suppression and 24-hour urine free cortisol tests have the highest specificity (97% and 91%, respectively) and the most evidence to support their use8,18 (Table 26–12,17–19 ). When the diagnosis remains uncertain after these initial studies, dynamic endocrine function tests should be performed, usually in consultation with an endocrinologist.
A higher serum prolactin level (250 mcg per L [10,870 pmol per L] or more) suggests a prolactinoma over other causes of hyperprolactinemia (e.g., hypothyroidism, medications, non–prolactin-secreting pituitary adenomas, pregnancy, renal failure).9,17 A serum prolactin level greater than 500 mcg per L (21,739 pmol per L) is diagnostic for a macroprolactinoma (98% specificity), although only about one-third of persons with prolactinoma have levels that high (35% sensitivity).17 Non–prolactin-secreting pituitary tumors may cause moderate elevations in prolactin through compression of the pituitary stalk, resulting in loss of inhibition of prolactin release; this is termed the stalk effect.6,17
NEUROLOGIC SYMPTOMS
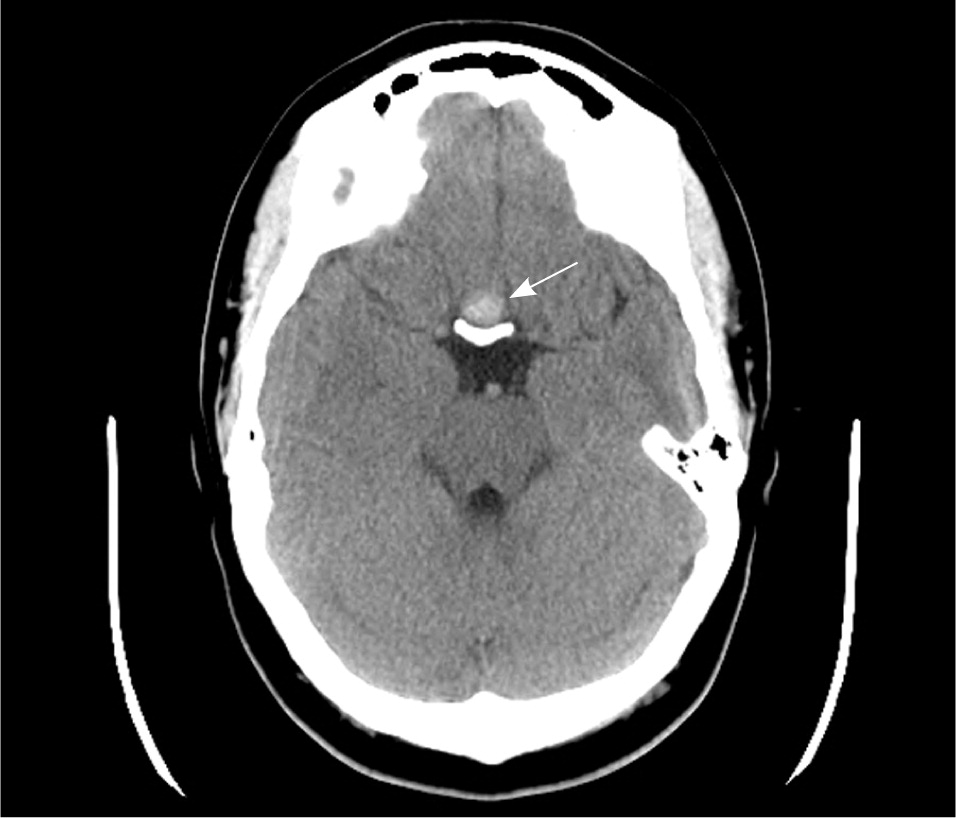
If a pituitary mass is suspected, MRI is the best initial imaging study.7 MRI is 61% to 72% sensitive and 88% to 90% specific for sellar masses.20,21 The study should be done with and without gadolinium enhancement. CT is limited by its inability to precisely image the optic chiasm. If an MRI is contraindicated or unavailable, CT done with thin sections (1.5 mm or less) and in a coronal plane will improve imaging of the pituitary region22 (Figures 2 and 3).
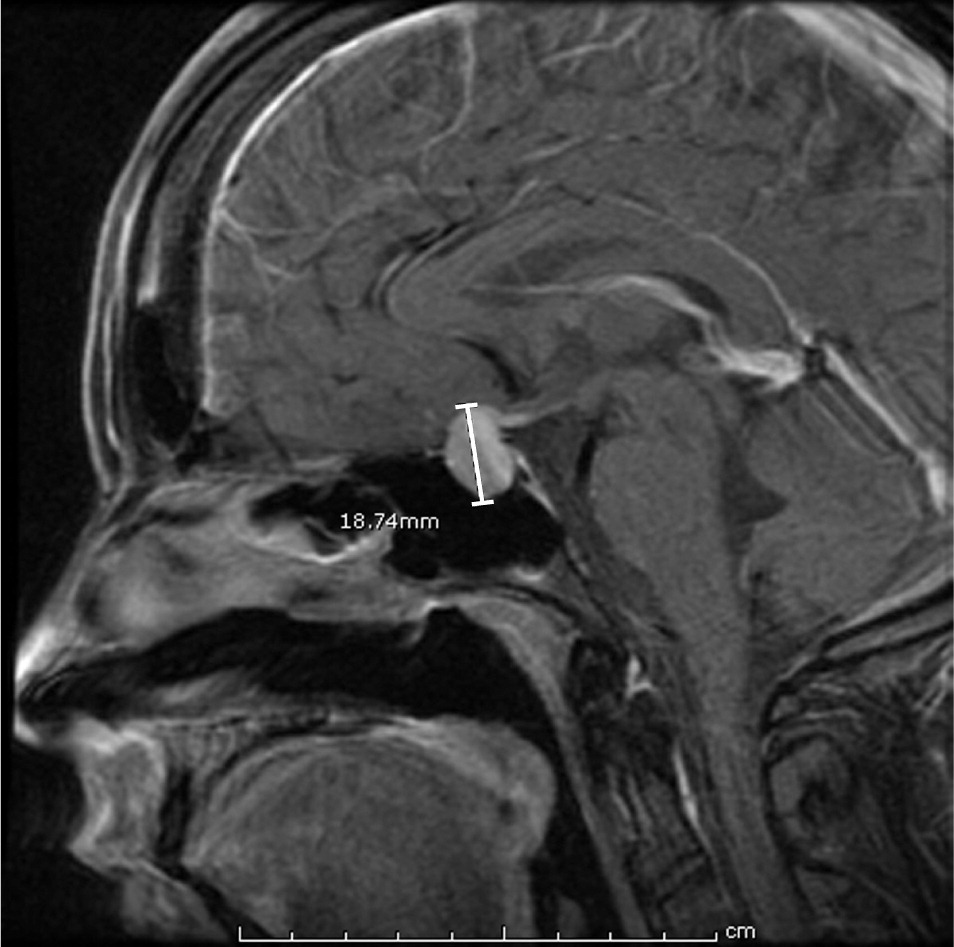
If a patient presents with visual symptoms, or if imaging reveals impingement on the optic nerve, the patient should be referred for formal visual field testing and a complete ophthalmologic examination.7 Early referral for visual field testing should be considered even in the absence of presenting symptoms, because unrecognized deficits occur in approximately 10% of incidentally discovered pituitary adenomas.15,16
INCIDENTALOMA
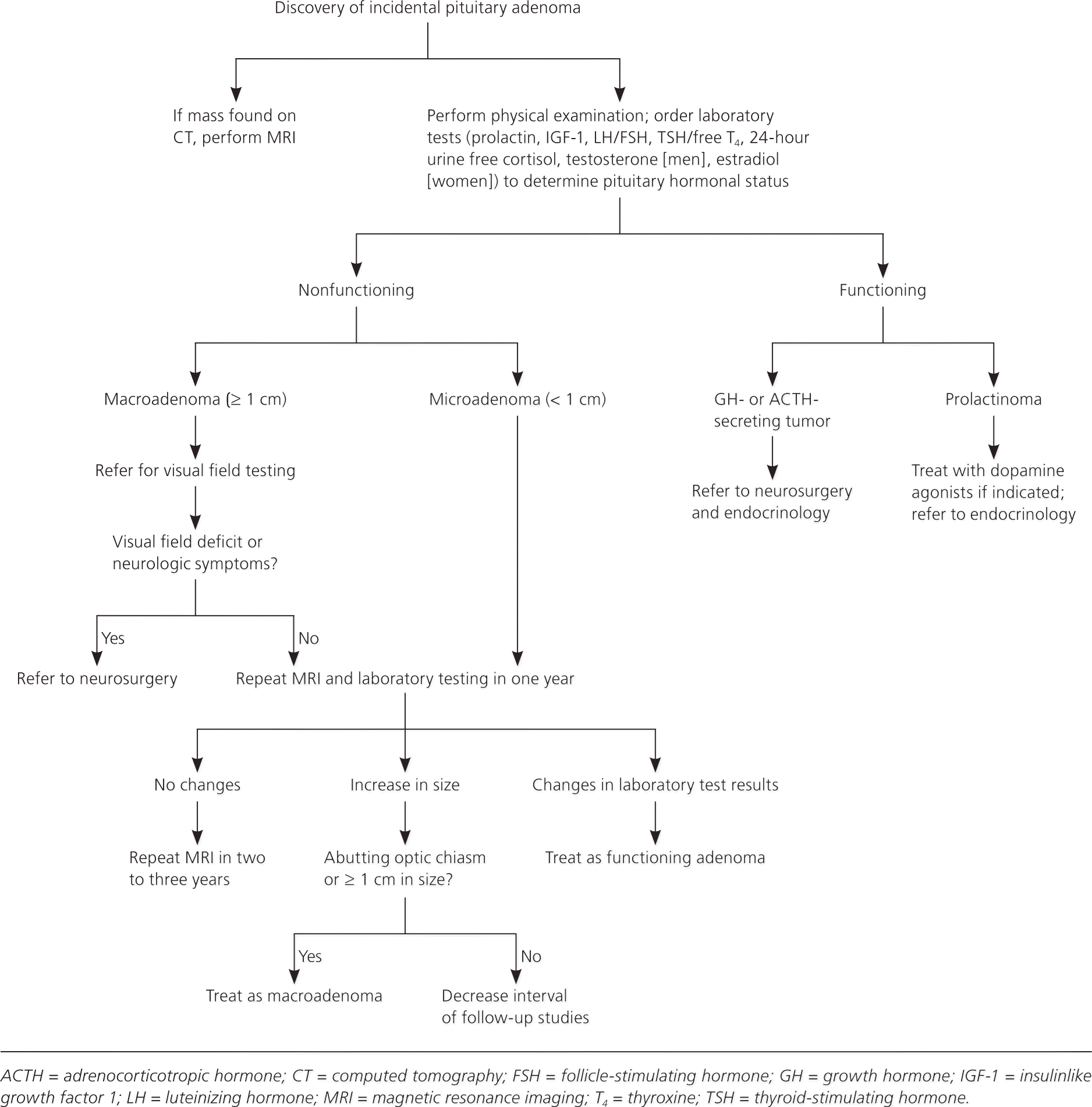
When a pituitary mass is discovered incidentally, diagnostic studies should be geared toward determining if the mass is functioning or nonfunctioning (Figure 47 ). There is debate over the extent of laboratory assessment warranted in the evaluation of incidentalomas, especially microadenomas. The Endocrine Society's 2011 clinical practice guidelines for pituitary incidentalomas recommend a complete assessment of pituitary function, even if the patient is asymptomatic.7 The basis for this recommendation stems from the effectiveness of prolactinoma treatment and the value of identifying growth hormone– and ACTH-secreting adenomas early to avoid associated long-term morbidity. Most patients do not require surgical intervention, although prospective biochemical monitoring and repeated imaging are recommended by expert panel guidelines.7 Clinical trials of the best approach to monitoring these patients have not been done.
Therapy
The majority of prolactinomas can be managed medically with dopamine agonists. Dopamine agonists approved for use in the United States are bromocriptine (Parlodel) and cabergoline. By inhibiting the release of prolactin from the anterior pituitary, these medications resolve hyperprolactinemia symptoms, reduce tumor size, and often restore reproductive function.9,17,28 Several controlled trials have shown improved effectiveness and better patient tolerance of cabergoline over bromocriptine. Patient-oriented outcomes such as quicker restoration of normal vision, faster return of regular menses, and fewer gastrointestinal adverse effects were noted in those using cabergoline.23,24
The most common adverse effects of dopamine agonists are nausea, vomiting, and fatigue. Long-term use of high-dose ergoline-derived dopamine agonists for Parkinson disease increases the risk of cardiac valvular regurgitation. However, this same association has not been found with use of short-term, lower-dose agents in the treatment of prolactinomas.29
Although dopamine agonists are not approved for use during pregnancy, both bromocriptine and cabergoline are thought to be safe.28 Because of the greater body of published evidence on bromocriptine, it is the recommended dopamine agonist for initiation during pregnancy.9,28 Expert consultation is advised for all women with a prolactinoma who intend to become pregnant.
Medical management of growth hormone– and ACTH-secreting tumors is less effective than for prolactinomas, and surgery via transsphenoidal resection is the preferred treatment.7,25,30 Somatostatin analogues such as octreotide (Sandostatin) and lanreotide (Somatuline) inhibit growth hormone secretion and somatotroph proliferation. They decrease the size of growth hormone–secreting tumors and the symptoms of growth hormone excess. The growth hormone receptor antagonist pegvisomant (Somavert) reduces the production of IGF-1, which is primarily responsible for the symptoms of acromegaly.26 Medications that decrease or halt steroidogenesis at the adrenal gland (ketoconazole, metyrapone [Metopirone], mitotane [Lysodren], and mifepristone [Mifeprex]) are used to decrease symptoms in patients with ACTH-secreting tumors, and for palliation in persons unable to tolerate surgery (Table 39,10,23–27 ), but they have no effect on the tumor itself or in restoration of normal pituitary function.27 Radiation and radiosurgery can decrease tumor size and enhance endocrine functioning, especially when used to treat post-resection residual adenoma.27,31,32
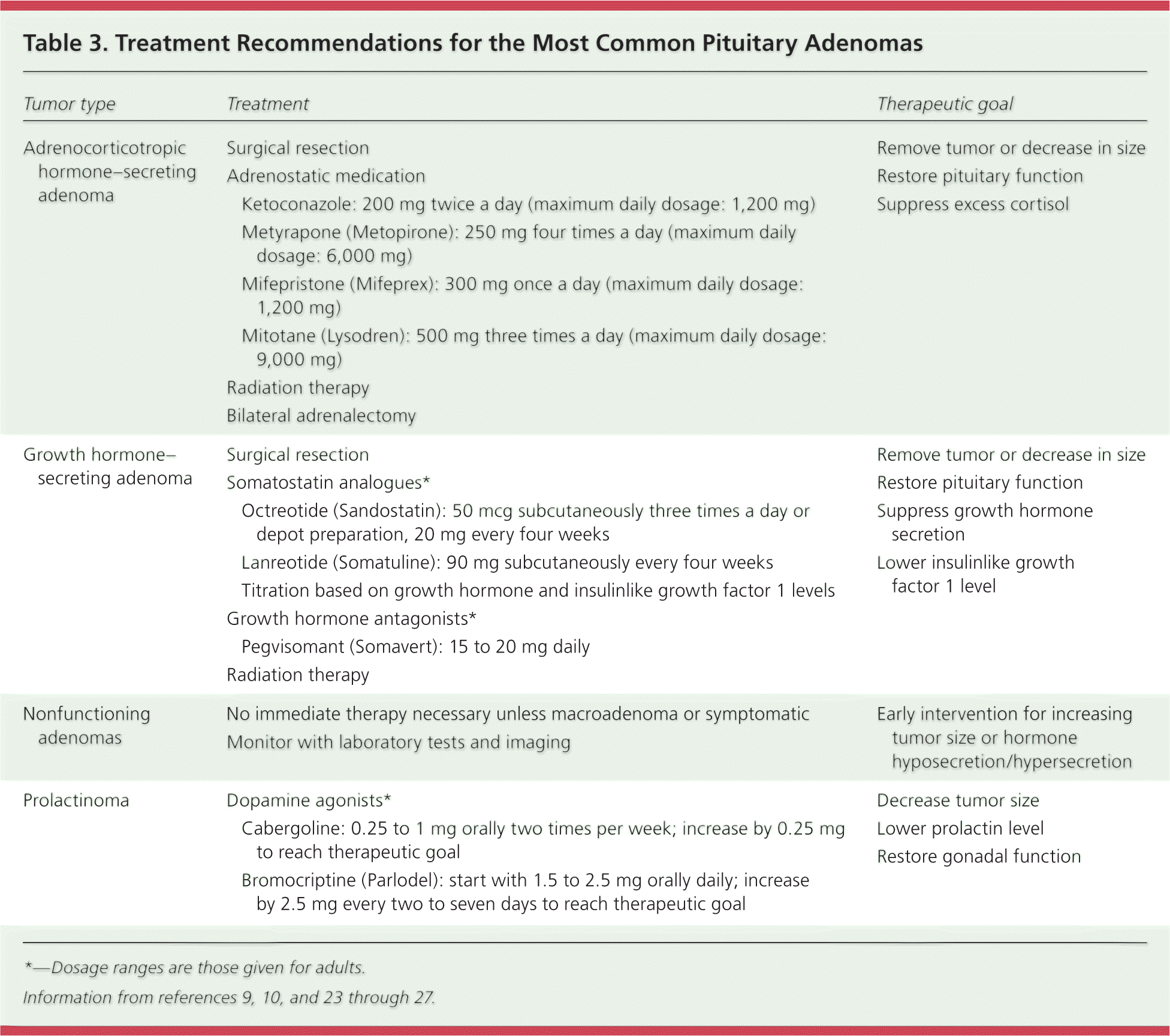
| Tumor type | Treatment | Therapeutic goal | |
|---|---|---|---|
| Adrenocorticotropic hormone–secreting adenoma | Surgical resection |
| |
| Adrenostatic medication | |||
| Ketoconazole: 200 mg twice a day (maximum daily dosage: 1,200 mg) | |||
| Metyrapone (Metopirone): 250 mg four times a day (maximum daily dosage: 6,000 mg) | |||
| Mifepristone (Mifeprex): 300 mg once a day (maximum daily dosage: 1,200 mg) | |||
| Mitotane (Lysodren): 500 mg three times a day (maximum daily dosage: 9,000 mg) | |||
| Radiation therapy | |||
| Bilateral adrenalectomy | |||
| Growth hormone–secreting adenoma | Surgical resection |
| |
| Somatostatin analogues* | |||
| Octreotide (Sandostatin): 50 mcg subcutaneously three times a day or depot preparation, 20 mg every four weeks | |||
| Lanreotide (Somatuline): 90 mg subcutaneously every four weeks | |||
| Titration based on growth hormone and insulinlike growth factor 1 levels | |||
| Growth hormone antagonists* | |||
| Pegvisomant (Somavert): 15 to 20 mg daily | |||
| Radiation therapy | |||
| Nonfunctioning adenomas | No immediate therapy necessary unless macroadenoma or symptomatic |
| |
| Monitor with laboratory tests and imaging | |||
| Prolactinoma | Dopamine agonists* |
| |
| Cabergoline: 0.25 to 1 mg orally two times per week; increase by 0.25 mg to reach therapeutic goal | |||
| Bromocriptine (Parlodel): start with 1.5 to 2.5 mg orally daily; increase by 2.5 mg every two to seven days to reach therapeutic goal | |||
Nonfunctioning microadenomas and microprolactinomas in asymptomatic patients do not require immediate treatment. However, a small percentage of these tumors will increase in size or cause new pituitary dysfunction, and therefore warrant monitoring.16,33 No clinical trials have been done to compare a conservative approach with early therapy, so management and monitoring intervals are based on expert opinion. An expert guideline recommends a repeat MRI in 12 months and, if there is no increase in size, lengthening the imaging interval to two to three years.7 Biochemical testing via an endocrine panel is not recommended unless the tumor enlarges or the patient develops symptoms (Figure 47 ). Measurement of prolactin alone may also be an effective strategy to monitor microadenomas; a cost analysis found this test to have the lowest cost per quality-adjusted life-year.34
Data Sources: A PubMed search was completed in Clinical Queries using the key terms pituitary adenoma, prolactinoma, pituitary incidentaloma, Cushing's disease, and acromegaly in both diagnosis and treatment. The search included systematic reviews, meta-analyses, clinical practice guidelines, and clinical studies. We also used the National Guideline Clearinghouse, Essential Evidence Plus, the Cochrane database, UpToDate, and Google Scholar. Search dates: June 2011 and January 2012.
