
Am Fam Physician. 2014;89(2):89-90
Author disclosure: No relevant financial affiliations.
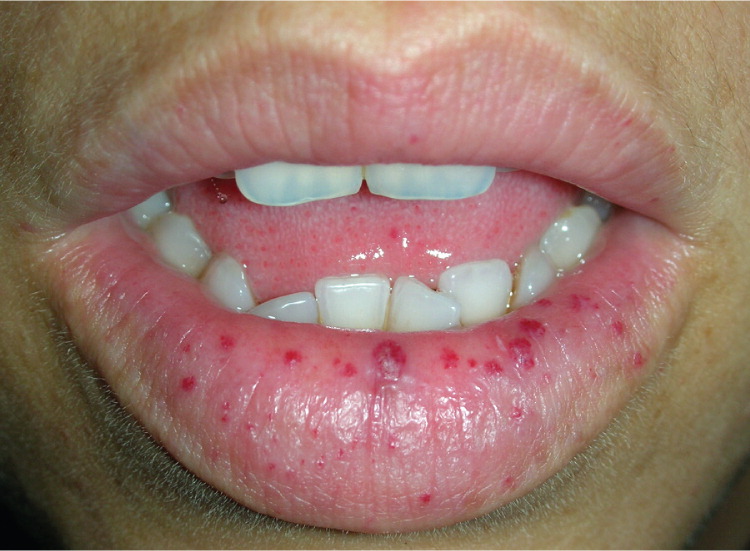
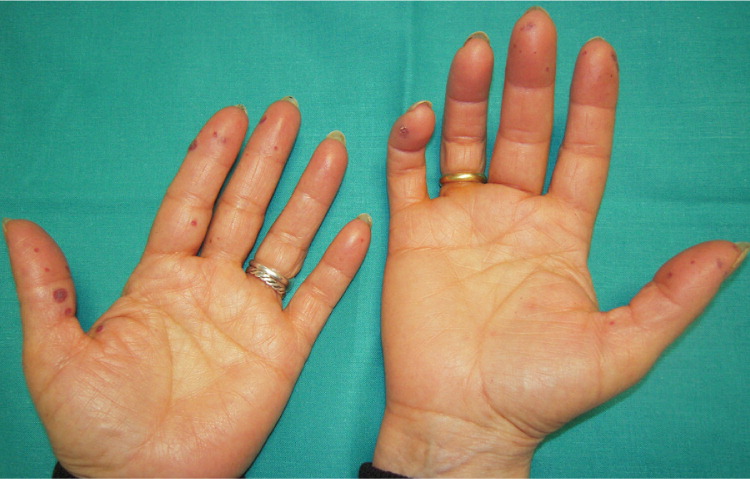
A 29-year-old woman presented with an eight-month history of progressive fatigue and recurrent epistaxis. She reported that her father and her grandmother also had a history of nosebleeds. The physical examination revealed telangiectasias on her lower lip, tongue, and fingers (Figures 1 and 2). Laboratory tests showed severe hypochromic microcytic anemia, with a hemoglobin level of 4.1 g per dL (41 g per L; normal range: 12 to 14 g per dL [120 to 140 g per L]).
Question
Discussion
The correct answer is C: hereditary hemorrhagic telangiectasia. This autosomal dominant vascular disorder, which is also referred to as Osler-Weber-Rendu disease, has a variety of clinical manifestations, including epistaxis, gastrointestinal bleeding, iron deficiency anemia, and mucocutaneous telangiectasias. Epidemiologic studies suggest the prevalence is one in 5,000 to 8,000 persons in most populations.1
Spontaneous, recurrent epistaxis from telangiectasia of the nasal mucosa is the most common clinical manifestation of hereditary hemorrhagic telangiectasia. Although some patients experience minimal or only occasional episodes, many have daily bleeds that can lead to iron deficiency anemia.2 Mucocutaneous telangiectasias occur in about 75% of patients with the condition, typically presenting after childhood and increasing in size and number with age. They are most common on the lips, tongue, buccal mucosa, and fingertips. Pulmonary, hepatic, and cerebral arteriovenous malformations are possible and may lead to embolic stroke, brain abscess, and hemorrhagic stroke.
A hereditary hemorrhagic telangiectasia diagnosis is clinical and based on the Curacao criteria: spontaneous, recurrent epistaxis; multiple mucocutaneous telangiectasias; visceral involvement (e.g., gastrointestinal, pulmonary, cerebral, or hepatic arteriovenous malformations); and a first-degree relative with the disease.3,4 The diagnosis can be made if three or four criteria are present; the diagnosis is suspected if two are present, and is unlikely if one or none is present. Formal genetic testing can confirm the disease.
Blue rubber bleb nevus syndrome, or Bean syndrome, is a rare disorder. Patients with this condition present with venous malformations of the skin and internal viscera appearing as multiple, blue to blue-gray macules, papules, and nodules.5
CREST (calcinosis cutis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia) syndrome may lead to fibrotic skin changes, often involving the fingers, hands, and face. Macular telangiectasias, usually on the lips and palms, are matted or squared-off, as opposed to the raised lesions associated with hereditary hemorrhagic telangiectasia. Capillary abnormalities in the proximal nail fold, sclerodactyly, digital pitting scars, and loss of substance from the finger pad are also distinguishing characteristics.6
Peutz-Jeghers syndrome is a rare genetic disorder characterized by melanotic macules, gastrointestinal polyps, and increased cancer risk. Melanotic macules are typically flat, blue-gray to brown, 1- to 5-mm spots.7

| Condition | Characteristics |
|---|---|
| Blue rubber bleb nevus syndrome | Venous malformations of the skin and internal viscera appearing as multiple, blue to blue-gray macules, papules, and nodules |
| CREST syndrome | Telangiectatic matted or squared-off macules on the lips and palms; capillary abnormalities in the proximal nail fold; sclerodactyly; digital pitting scars; loss of substance from the finger pad |
| Hereditary hemorrhagic telangiectasia | Autosomal dominant vascular disorder; manifestations include epistaxis, gastrointestinal bleeding, iron deficiency anemia, and mucocutaneous telangiectasias |
| Peutz-Jeghers syndrome | Melanotic macules; gastrointestinal polyps; increased cancer risk |
