
A more recent article on the evaluation of short and tall stature in children is available.
Am Fam Physician. 2015;92(1):43-50
Author disclosure: No relevant financial affiliations.
Short stature is defined as a height more than two standard deviations below the mean for age (less than the 3rd percentile). Tall stature is defined as a height more than two standard deviations above the mean for age (greater than the 97th percentile). The initial evaluation of short and tall stature should include a history and physical examination, accurate serial measurements, and determination of growth velocity, midparental height, and bone age. Common normal variants of short stature are familial short stature, constitutional delay of growth and puberty, and idiopathic short stature. Pathologic causes of short stature include chronic diseases; growth hormone deficiency; and genetic disorders, such as Turner syndrome. Tall stature has the same prevalence as short stature, but it is a much less common reason for referral to subspecialty care. Common causes of tall stature include familial tall stature, obesity, Klinefelter syndrome, Marfan syndrome, and precocious puberty. Although most children with short or tall stature have variants of normal growth, children who are more than three standard deviations from the mean for age are more likely to have underlying pathology. Evaluation for pathologic etiologies is guided by history and physical examination findings.
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| History and physical examination findings should guide further evaluation for pathologic causes of short and tall stature. | C | 1–3 |
| World Health Organization growth charts should be used for children younger than two years, and the Centers for Disease Control and Prevention growth charts should be used for children older than two years. | C | 5 |
| Midparental height growth velocity should be calculated to evaluate a child's growth vs. potential height. | C | 6–8 |
| Bone age should be compared with chronologic age to help narrow the differential diagnosis of short or tall stature. | C | 5, 10, 11 |
Evaluation of Growth
The first step in the evaluation of a child with suspected short or tall stature is to obtain accurate measurements and plot them on the appropriate growth chart. For infants and toddlers, weight, length, and head circumference should be plotted on a growth curve at every visit. For patients two to 20 years of age, weight, height, and body mass index should be plotted. Length should be measured using a horizontal rule in children younger than two years, and height should be measured using a wall-mounted stadiometer in children older than two years. Because children grow in spurts, two measurements at least three to six months apart, and preferably six to 12 months apart, are needed to accurately determine growth velocity.4
The Centers for Disease Control and Prevention (CDC) and the American Academy of Pediatrics recommend using the World Health Organization (WHO) growth charts for children younger than two years and the CDC growth charts for children older than two years.5 The CDC growth charts are a population-based reference that include data from bottle-fed and breastfed infants. Because the WHO growth charts are based on an international study of exclusively breastfed infants raised in optimal nutritional conditions, they are less likely to incorrectly identify breastfed infants as underweight. The CDC and WHO growth charts are available at http://www.cdc.gov/growthcharts/ and http://www.who.int/childgrowth/standards/en/.
A newborn's size and growth are a result of the intrauterine environment, and growth hormone does not play a major role. Between six and 18 months of age, children exhibit catch-up or catch-down growth until they reach their genetically determined growth curve based on midparental height. By two years of age, growth hormone plays a predominant role. At this stage, children should track along a percentile, and variation should stay within two large bands on the growth chart. In adolescence, growth is affected by the onset of puberty, and sex hormones become the predominant factor in growth.
Variation from this normal pattern of growth may be a sign of pathologic conditions. Although most children with short or tall stature do not have a pathologic condition, extremes of height, especially beyond three standard deviations, require further workup.
MIDPARENTAL AND PROJECTED HEIGHT
Calculating the midparental height (Table 1) is an important part of the evaluation because most short or tall children have short or tall parents. Projected height can be estimated by projecting the current growth curve to adulthood in children with normal bone age, or by using a bone age atlas in those with delayed bone age. Most children will have a projected adult height within 10 cm (4 in), or two standard deviations, of their midparental height. A projected height that differs from the midparental height by more than 10 cm suggests a possible pathologic condition. Short or tall parents may themselves have a pathologic reason for their height, especially if they are more than two standard deviations from the adult norm.6–8

| Girls | |
| [Paternal height (cm) – 13 cm + maternal height (cm)] ÷ 2 | |
| or | |
| [Paternal height (in) – 5 in + maternal height (in)] ÷ 2 | |
| Boys | |
| [Paternal height (cm) + 13 cm + maternal height (cm)] ÷ 2 | |
| or | |
| [Paternal height (in) + 5 in + maternal height (in)] ÷ 2 |
Growth velocity is a measurement of growth rate. Children with normal variants of height tend to have a normal growth velocity (5 cm [2 in] per year for children between five years of age and puberty) after catch-up or catch-down growth. A growth velocity that is less than normal should prompt further investigation. Table 2 includes normal growth velocity by age.1,9

| Age | Growth velocity per year |
|---|---|
| Birth to 12 months | 23 to 27 cm (9.06 to 10.63 in) |
| 12 months to 1 year | 10 to 14 cm (3.94 to 5.51 in) |
| 2 to 3 years | 8 cm (3.15 in) |
| 3 to 5 years | 7 cm (2.76 in) |
| 5 years to puberty | 5 to 6 cm (1.97 to 2.36 in) |
| Puberty | Girls: 8 to 12 cm (3.15 to 4.72 in) |
| Boys: 10 to 14 cm (3.94 to 5.51 in) |
BONE AGE
Bone age should be compared with chronologic age to narrow the differential diagnosis of short stature.5,10,11 The traditional method compares a plain radiograph of the left wrist and hand to a database of norms, although various methods are now available.10–12 Children with normal variations of growth may have advanced or delayed bone age, but a bone age that is more than two standard deviations from the mean for age is likely due to a pathologic condition.
Short Stature
Short stature is defined as a height more than two standard deviations below the mean for age, or less than the 3rd percentile. Idiopathic short stature is defined as a height less than two standard deviations below the mean for age without a known etiology.
Most children with short stature have normal variants such as familial short stature, constitutional delay of growth and puberty, or idiopathic short stature. Approximately 5% of children referred for evaluation of short stature have an identifiable pathologic cause.13 The most common etiologies are growth hormone deficiency, hypothyroidism, celiac disease, and Turner syndrome. Other causes include renal, hepatic, and gastrointestinal diseases, and other genetic syndromes.10–15
EVALUATION
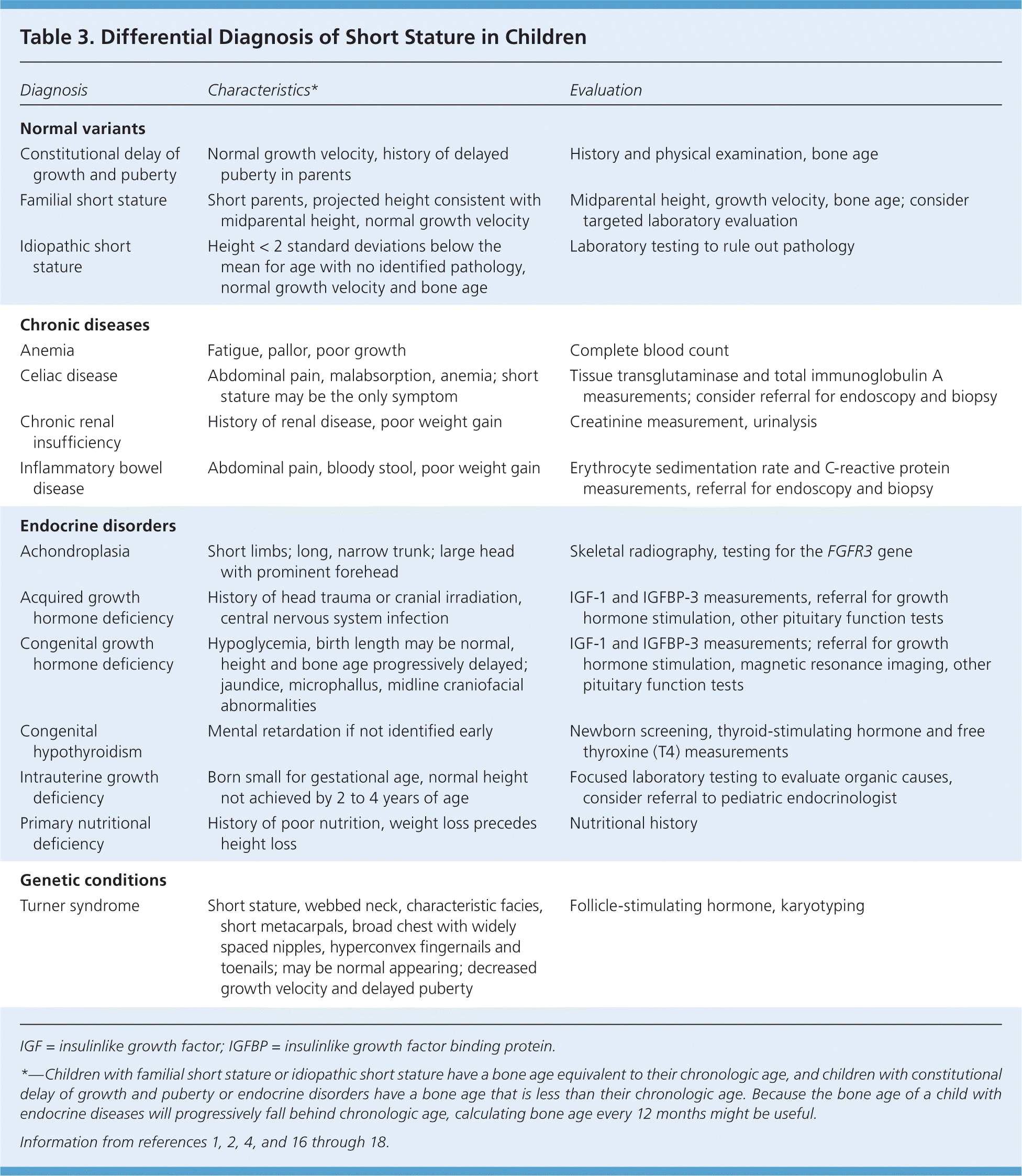
The initial evaluation of short stature (Figure 1) should include a history and physical examination, accurate growth assessment, calculation of the growth velocity and midparental height, and radiography to evaluate bone age.16 Drugs known to cause short stature include steroids (chronic use), attention-deficit/hyperactivity disorder medications, and anticonvulsants. Dysmorphic characteristics suggest a genetic disorder, whereas midline defects suggest an abnormality of the growth hormone axis. Table 3 includes the differential diagnosis of short stature.1,2,4,16–18
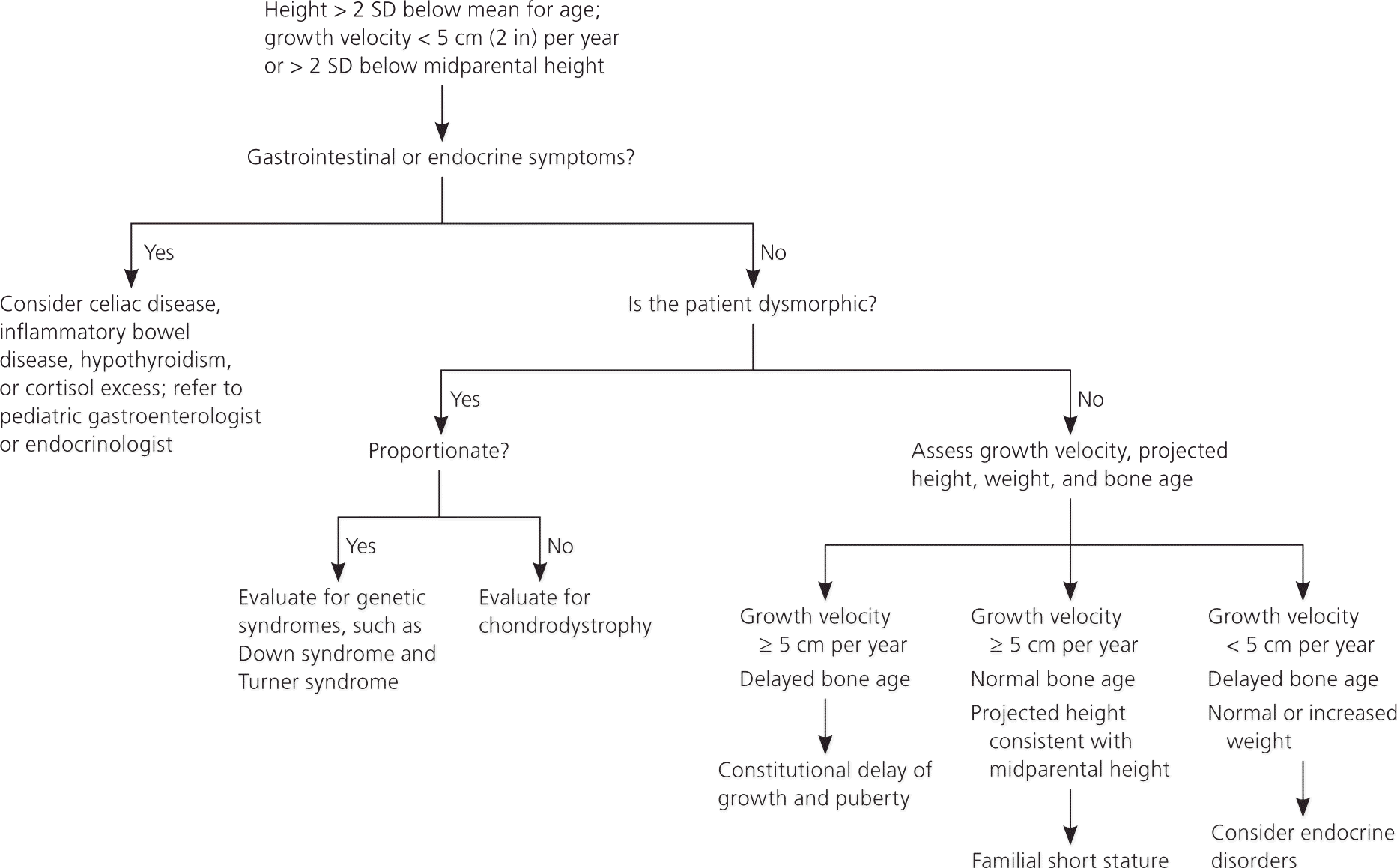
| Diagnosis | Characteristics* | Evaluation |
|---|---|---|
| Normal variants | ||
| Constitutional delay of growth and puberty | Normal growth velocity, history of delayed puberty in parents | History and physical examination, bone age |
| Familial short stature | Short parents, projected height consistent with midparental height, normal growth velocity | Midparental height, growth velocity, bone age; consider targeted laboratory evaluation |
| Idiopathic short stature | Height < 2 standard deviations below the mean for age with no identified pathology, normal growth velocity and bone age | Laboratory testing to rule out pathology |
| Chronic diseases | ||
| Anemia | Fatigue, pallor, poor growth | Complete blood count |
| Celiac disease | Abdominal pain, malabsorption, anemia; short stature may be the only symptom | Tissue transglutaminase and total immunoglobulin A measurements; consider referral for endoscopy and biopsy |
| Chronic renal insufficiency | History of renal disease, poor weight gain | Creatinine measurement, urinalysis |
| Inflammatory bowel disease | Abdominal pain, bloody stool, poor weight gain | Erythrocyte sedimentation rate and C-reactive protein measurements, referral for endoscopy and biopsy |
| Endocrine disorders | ||
| Achondroplasia | Short limbs; long, narrow trunk; large head with prominent forehead | Skeletal radiography, testing for the FGFR3 gene |
| Acquired growth hormone deficiency | History of head trauma or cranial irradiation, central nervous system infection | IGF-1 and IGFBP-3 measurements, referral for growth hormone stimulation, other pituitary function tests |
| Congenital growth hormone deficiency | Hypoglycemia, birth length may be normal, height and bone age progressively delayed; jaundice, microphallus, midline craniofacial abnormalities | IGF-1 and IGFBP-3 measurements; referral for growth hormone stimulation, magnetic resonance imaging, other pituitary function tests |
| Congenital hypothyroidism | Mental retardation if not identified early | Newborn screening, thyroid-stimulating hormone and free thyroxine (T4) measurements |
| Intrauterine growth deficiency | Born small for gestational age, normal height not achieved by 2 to 4 years of age | Focused laboratory testing to evaluate organic causes, consider referral to pediatric endocrinologist |
| Primary nutritional deficiency | History of poor nutrition, weight loss precedes height loss | Nutritional history |
| Genetic conditions | ||
| Turner syndrome | Short stature, webbed neck, characteristic facies, short metacarpals, broad chest with widely spaced nipples, hyperconvex fingernails and toenails; may be normal appearing; decreased growth velocity and delayed puberty | Follicle-stimulating hormone, karyotyping |
If the initial evaluation suggests a genetic, endocrine, or gastrointestinal disorder, laboratory testing should be performed (Table 4).1,3,13,14,16,19,20 In an asymptomatic child with short stature, an evaluation of the growth curve may provide clues to the underlying pathology. Underweight in a child with short stature suggests a systemic illness or malnutrition, whereas overweight suggests an endocrine disorder.2,21

| Test | Indication |
|---|---|
| Short stature | |
| Complete blood count | Anemia |
| Comprehensive metabolic panel | Hepatic and renal diseases |
| Erythrocyte sedimentation rate, C-reactive protein | Inflammatory bowel disease |
| Follicle-stimulating hormone, karyotyping | Turner syndrome |
| Insulinlike growth factor 1* | Growth hormone deficiency |
| Thyroid-stimulating hormone, free thyroxine (T4) | Hypothyroidism |
| Tissue transglutaminase and total immunoglobulin A | Celiac disease |
| Urinalysis | Renal disease |
| Tall stature | |
| Insulinlike growth factor 1* | Growth hormone excess |
| Karyotyping | Klinefelter syndrome (XXY) |
| Serum luteinizing hormone, follicle-stimulating hormone, testosterone | Precocious puberty |
| Thyroid-stimulating hormone, free thyroxine (T4) | Hyperthyroidism |
Different causes of short stature tend to fall within identifiable growth patterns, and a review of a child's growth curve and bone age should guide further evaluation. Children with familial short stature or idiopathic short stature have a bone age equivalent to their chronologic age, and children with constitutional delay of growth and puberty or endocrine disorders have a bone age that is less than their chronologic age. Because the bone age of a child with endocrine diseases will progressively fall behind chronologic age, calculating bone age every 12 months might be useful to differentiate constitutional delay of growth from endocrine diseases.1
If findings from the initial evaluation do not suggest a diagnosis, laboratory testing may be performed (Table 4).1,3,13,14,16,19,20 A retrospective study found that a complete laboratory evaluation of an asymptomatic child with idiopathic short stature is low yield and expensive. The two diseases that were most often identified in the studied cohort were celiac disease and an abnormality of the growth hormone axis.3 If history and physical examination findings do not suggest a cause, a complete blood count, comprehensive metabolic panel, and measurement of bone age, insulinlike growth factor 1, and insulinlike growth factor binding protein 3 might be useful to screen for chronic disease and growth hormone deficiency. Karyotyping in girls might also be reasonable because short stature and delayed puberty may be the only symptoms in some girls with Turner syndrome.
REFERRAL

| Children with intrauterine growth retardation who do not catch up to the growth curve by 2 years of age |
| Height more than 3 standard deviations below the mean for age |
| Growth velocity < 5 cm (2 in) per year |
| No onset of puberty by 14 years of age for boys or 13 years of age for girls |
| Projected height more than 2 standard deviations (10 cm [4 in]) below the midparental height |
| Bone age more than 2 standard deviations below chronologic age |
| Diagnosis of conditions approved for recombinant growth hormone therapy* |
SELECTED CAUSES
Constitutional Delay of Growth and Puberty. Children with this condition are born appropriate for gestational age, but will then fall to the 3rd percentile for height during catch-down growth. After this period, growth velocity will be normal and bone age delayed.22 Children with this condition have delayed onset of puberty, resulting in a normal adult height.
Growth Hormone Deficiency. This condition may be congenital or acquired, and has an incidence of one in 3,000 to 9,000 children.13 A history of head trauma, central nervous system infection, birth trauma, or cranial irradiation may suggest an acquired cause of growth hormone deficiency. Most infants with the congenital form are normal size at birth, but may have episodes of hypoglycemia or prolonged jaundice. Physical examination may reveal microphallus or midline craniofacial abnormalities. A child whose growth is initially normal but then falls progressively further off the growth curve may have growth hormone deficiency. Typically, children with this condition have a delayed bone age with a preserved or increased weight for age. The diagnosis can be made by a decreased insulinlike growth factor 1 or insulinlike growth factor binding protein 3, followed by negative growth hormone provocation test results.23
Small for Gestational Age. Infants born small for gestational age typically have catch-up growth in the first 24 months, but 10% have a final height more than two standard deviations below the mean for age.24 Children who do not have catch-up growth within the first six months or whose height is not within two standard deviations of the mean for age by two years of age may have a pathologic condition. It may take more than four years for a preterm infant who is born small for gestational age to attain a normal height.24
TREATMENT
Recombinant growth hormone is approved for a variety of conditions that cause short stature, including Turner syndrome, chronic renal failure, Prader-Willi syndrome, small for gestational age, Noonan syndrome, short stature homeobox-containing gene deficiency, and idiopathic short stature. It is administered through daily injections over several years. The injections are generally well tolerated, but rare adverse reactions have been reported. For children with idiopathic short stature, four years of treatment results in an increased height of 3.7 cm (1.46 in) and costs between $100,000 and $120,000.25,26
Oxandrolone (Oxandrin) is an oral anabolic steroid that has been shown to increase height velocity but has little effect on final height. Insulinlike growth factor has been used in children with insulinlike growth factor deficiency. Although aromatase inhibitors have been used in children with idiopathic short stature, long-term effectiveness and safety data are not available.27
Tall Stature
Tall stature is defined as a height more than two standard deviations above the mean for age (greater than the 97th percentile). Evaluation may also be needed in a child who has a normal height, but a projected height more than two standard deviations from the midparental height. Figure 2 is an algorithm for the evaluation of tall stature.19 Although the percentage of children with tall stature is equal to that of children with short stature, children with tall stature are much less likely to be referred to subspecialty care.
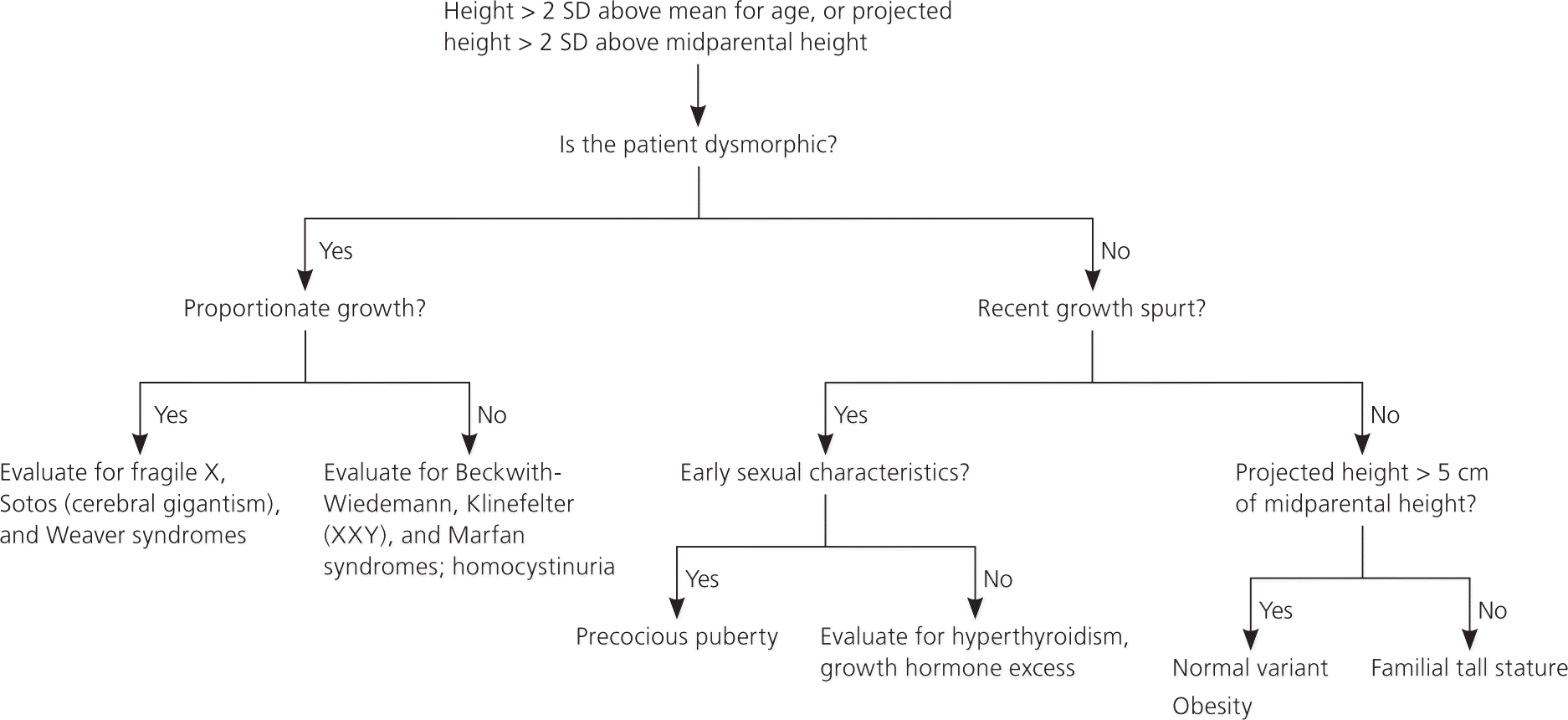
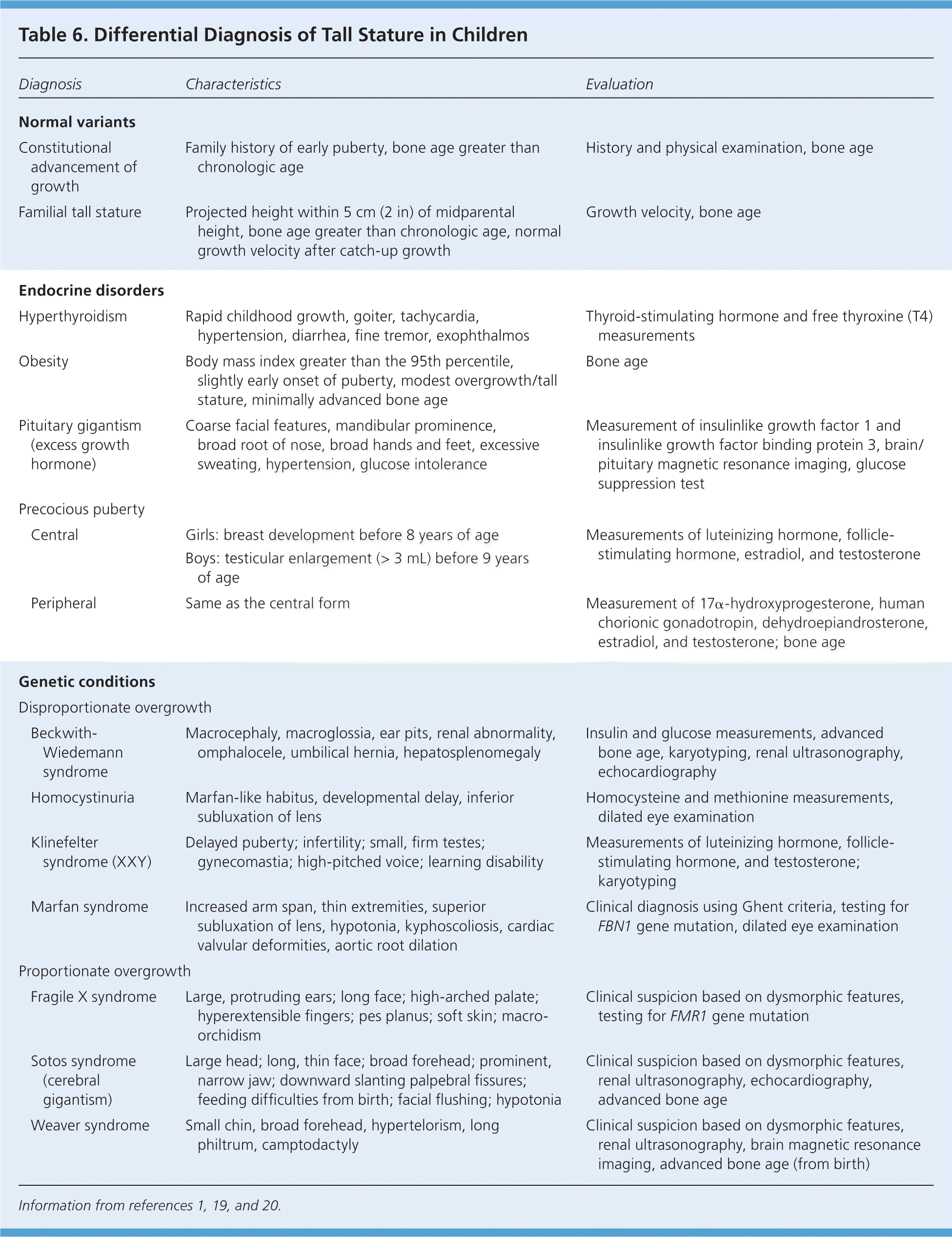
Table 6 includes the differential diagnosis of tall stature. Constitutional advancement of growth in tall children is the equivalent of constitutional delay of growth and puberty in short children.1,19,20 Children with constitutional advancement of growth have accelerated growth until two to four years of age and then track parallel to the growth curve. Puberty usually occurs early, leading to a near-normal height.19
| Diagnosis | Characteristics | Evaluation | |
|---|---|---|---|
| Normal variants | |||
| Constitutional advancement of growth | Family history of early puberty, bone age greater than chronologic age | History and physical examination, bone age | |
| Familial tall stature | Projected height within 5 cm (2 in) of midparental height, bone age greater than chronologic age, normal growth velocity after catch-up growth | Growth velocity, bone age | |
| Endocrine disorders | |||
| Hyperthyroidism | Rapid childhood growth, goiter, tachycardia, hypertension, diarrhea, fine tremor, exophthalmos | Thyroid-stimulating hormone and free thyroxine (T4) measurements | |
| Obesity | Body mass index greater than the 95th percentile, slightly early onset of puberty, modest overgrowth/tall stature, minimally advanced bone age | Bone age | |
| Pituitary gigantism (excess growth hormone) | Coarse facial features, mandibular prominence, broad root of nose, broad hands and feet, excessive sweating, hypertension, glucose intolerance | Measurement of insulinlike growth factor 1 and insulinlike growth factor binding protein 3, brain/pituitary magnetic resonance imaging, glucose suppression test | |
| Precocious puberty | |||
| Central | Girls: breast development before 8 years of age | Measurements of luteinizing hormone, follicle-stimulating hormone, estradiol, and testosterone | |
| Boys: testicular enlargement (> 3 mL) before 9 years of age | |||
| Peripheral | Same as the central form | Measurement of 17α-hydroxyprogesterone, human chorionic gonadotropin, dehydroepiandrosterone, estradiol, and testosterone; bone age | |
| Genetic conditions | |||
| Disproportionate overgrowth | |||
| Beckwith-Wiedemann syndrome | Macrocephaly, macroglossia, ear pits, renal abnormality, omphalocele, umbilical hernia, hepatosplenomegaly | Insulin and glucose measurements, advanced bone age, karyotyping, renal ultrasonography, echocardiography | |
| Homocystinuria | Marfan-like habitus, developmental delay, inferior subluxation of lens | Homocysteine and methionine measurements, dilated eye examination | |
| Klinefelter syndrome (XXY) | Delayed puberty; infertility; small, firm testes; gynecomastia; high-pitched voice; learning disability | Measurements of luteinizing hormone, follicle-stimulating hormone, and testosterone; karyotyping | |
| Marfan syndrome | Increased arm span, thin extremities, superior subluxation of lens, hypotonia, kyphoscoliosis, cardiac valvular deformities, aortic root dilation | Clinical diagnosis using Ghent criteria, testing for FBN1 gene mutation, dilated eye examination | |
| Proportionate overgrowth | |||
| Fragile X syndrome | Large, protruding ears; long face; high-arched palate; hyperextensible fingers; pes planus; soft skin; macro-orchidism | Clinical suspicion based on dysmorphic features, testing for FMR1 gene mutation | |
| Sotos syndrome (cerebral gigantism) | Large head; long, thin face; broad forehead; prominent, narrow jaw; downward slanting palpebral fissures; feeding difficulties from birth; facial flushing; hypotonia | Clinical suspicion based on dysmorphic features, renal ultrasonography, echocardiography, advanced bone age | |
| Weaver syndrome | Small chin, broad forehead, hypertelorism, long philtrum, camptodactyly | Clinical suspicion based on dysmorphic features, renal ultrasonography, brain magnetic resonance imaging, advanced bone age (from birth) | |
TREATMENT
Intervention is usually not needed in children with tall stature. High-dose sex steroids have been used to promote growth plate closure, but use has decreased over the past 20 years because of adverse effects.28 Surgical destruction of the growth plates has also been performed, but this procedure is controversial. In patients with pituitary gigantism, octreotide (Sandostatin) and pegvisomant (Somavert) have been used to suppress the growth hormone.19
Data Sources: We searched PubMed, Agency for Healthcare Research and Quality, Cochrane Database of Systematic Reviews, and National Guidelines Clearinghouse. Search terms included short stature, tall stature, and growth hormone. We did online searches of The New England Journal of Medicine, Pediatrics, American Family Physician, Pediatrics in Review, and the British Medical Journal to identify additional relevant articles. The bibliographies of review articles and textbook chapters were also reviewed for original research articles. The Pediatric Endocrine Society website was searched for consensus statements and clinical guidelines. Search dates: June and December 2014, and March 2015.
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Army Medical Department or the U.S. Army Service at large.
