
Am Fam Physician. 2015;92(12):1069-1076A
Patient information: A handout on this topic is available at https://familydoctor.org/familydoctor/en/diseases-conditions/sickle-cell-disease.html.
Author disclosure: No relevant financial affiliations.
Family physicians are the primary and sometimes only health care resource for families affected by sickle cell disease. Recently published guidelines provide important recommendations for health maintenance, acute care, and monitoring of disease-modifying therapy in persons with this condition. This overview highlights some of the most important clinical activities that can and should be carried out in the community care setting. Children with sickle cell anemia should receive prophylactic penicillin from birth through at least five years of age, and all persons with sickle cell disease require vaccination to prevent invasive pneumococcal disease. Annual screening with transcranial Doppler ultrasonography is recommended for all children with sickle cell disease beginning at two years of age and continuing through adolescence to evaluate the risk of stroke and to initiate transfusion therapy in those at high risk. Vasoocclusive crises require immediate and adequate analgesia appropriate to the level of patient-reported pain. Antibiotics, hospitalization, and incentive spirometry are indicated for those with acute chest syndrome. There is strong evidence to support the promotion and use of hydroxyurea therapy in patients nine months and older who have sickle cell anemia because its use can decrease the frequency of vasoocclusive crises and acute chest syndrome with limited adverse effects.
Family physicians and family medical homes are essential to the care of children, adults, and families affected by sickle cell disease (SCD). Many of the approximately 100,000 U.S. residents who have SCD do not have regular or convenient access to comprehensive SCD centers. This summary of the recently published National Institutes of Health–sponsored SCD guidelines, Evidence-Based Management of Sickle Cell Disease, Expert Panel Report 2014,1 is intended to support, enhance, and expand the knowledge of basic aspects of care for patients with SCD. Specifically highlighted are several clinical actions to enhance preventive care, manage some of the most common acute and chronic complications of SCD, and initiate and monitor the two SCD-specific disease-modifying therapies, hydroxyurea and chronic blood transfusion therapy.
WHAT IS NEW ON THIS TOPIC: SICKLE CELL DISEASE
Offer hydroxyurea therapy to infants, children, and adolescents with sickle cell anemia regardless of clinical severity to reduce sickle cell disease–related complications.
Use an individualized prescribing and monitoring protocol or a sickle cell disease–specific protocol whenever possible to promote rapid, effective, and safe analgesic management and resolution of vasoocclusive crises in children and adults.
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Administer prophylactic oral penicillin (125 mg twice daily for children younger than three years; 250 mg twice daily for those three years and older) until at least five years of age in all children who are homozygous for sickle hemoglobin. | B | 9–11 |
| Screen children with sickle cell anemia annually with transcranial Doppler ultrasonography from two until at least 16 years of age. | A | 12–14 |
| Initiate rapid treatment with opioids in adults and children with sickle cell disease and a vasoocclusive crisis with severe pain. | B | 23–25 |
| Initiate hydroxyurea therapy in adults with sickle cell anemia who have one or more of the following: three or more moderate to severe pain crises in a 12-month period; daily sickle cell pain that affects quality of life; severe or recurrent acute chest syndrome; or severe symptomatic chronic anemia. | B | 1, 5, 32–34, 37 |
| Pregnant or breastfeeding women should discontinue hydroxyurea therapy. | C | 27 |
| Offer hydroxyurea therapy for infants nine to 42 months of age and for older children and adolescents with sickle cell anemia, regardless of clinical severity, to reduce sickle cell disease–related complications. | B | 35, 36, 38 |

| Recommendation | Sponsoring organization |
|---|---|
| Do not routinely transfuse patients with sickle cell disease for chronic anemia or uncomplicated pain crisis without an appropriate clinical indication. | American Society of Hematology |
Overview of SCD
SCD is a genetic disorder that results in the formation of sickled red blood cells (RBCs). Patients with SCD include those who are homozygous for sickle hemoglobin (HbSS, also called sickle cell anemia [SCA]) and those with one sickle hemoglobin gene plus a gene for another abnormal hemoglobin type (e.g., HbSβ+-thalassemia, HbSC). The recommendations in these guidelines do not apply to persons who have sickle cell trait (also called carriers [one gene for HbS and another for normal HbA]). SCD predominantly affects persons of African ancestry; a minority of affected persons are of Hispanic, Middle Eastern, or Asian Indian descent. Deoxygenated RBCs containing predominantly HbS develop a sickle or crescent shape, become inflexible, increase blood viscosity, and block or limit blood flow within limbs or organs.2 The process is further aggravated by abnormal interactions of these RBCs with leukocytes, platelets, vascular endothelium, and clotting factors,3,4 thus causing acute and chronic complications. The frequency and severity of complications can be reduced with hydroxyurea or blood transfusion therapy.5–8 However, both treatments are currently underutilized5–8 and are not commonly initiated or monitored by family physicians.
Caring for Patients with SCD in the Community Care Setting
SCREENING, PREVENTION, AND IMMUNIZATIONS
Many patients with SCD do not receive routine preventive care recommended by the U.S. Preventive Services Task Force and immunizations recommended by the Advisory Committee on Immunization Practices. Persons with SCD also benefit from specialized condition-specific preventive strategies. Children with SCA are at increased risk of invasive pneumococcal disease. Therefore, in addition to the recommended pneumococcal vaccinations for all infants and children, those with SCA should receive prophylactic oral penicillin (125 mg twice daily for children younger than three years; 250 mg twice daily for those three years and older) once the diagnosis is established; this regimen should be continued until at least five years of age.9–11
Persons with SCD are also at increased risk of vascular complications, particularly stroke. All persons with SCA should be screened annually with transcranial Doppler ultrasonography (TCD) beginning at two years of age and continuing until at least 16 years of age.12–14 TCD measures and reports the average velocity of blood flow through the internal carotid and proximal middle cerebral arteries. A TCD finding of 200 cm per second or greater correlates with a 10% annual increase in stroke risk.7 When TCD findings are marginal or conditional (170 to 199 cm per second) or elevated (200 cm per second or greater), the child is considered a candidate for blood transfusion therapy to prevent stroke and should be referred for comanagement with a subspecialist.15,16

| Prevention of invasive pneumococcal disease |
| Administer prophylactic oral penicillin (125 mg for children younger than 3 years; 250 mg for children 3 years and older) twice daily until 5 years of age in all children with SCA. (SOR: strong, based on moderate-quality evidence) |
| Ensure that all persons with SCD have been vaccinated against Streptococcus pneumoniae. (SOR: strong, based on moderate-quality evidence) |
| Discontinue prophylactic penicillin in children with SCA at 5 years of age unless they have had a splenectomy or invasive pneumococcal disease. Ensure completion of pneumococcal vaccination series before discontinuation. (SOR: weak, based on moderate-quality evidence) |
| Ensure that children 6 to 18 years of age who have functional or anatomic asplenia have received at least 1 dose of 13-valent pneumococcal conjugate vaccine. (SOR: adapted from the Advisory Committee on Immunization Practices) |
| Screening for ischemic retinopathy |
| Refer patients with SCD to an ophthalmologist for an annual dilated retinal examination beginning at 10 years of age. (SOR: strong, based on low-quality evidence) |
| Rescreen persons with normal dilated retinal examination findings at 1- to 2-year intervals. (SOR: consensus) |
| Screening for renal disease |
| Begin annual screening of persons with SCD for microalbuminuria and proteinuria with spot urine testing by 10 years of age. (SOR: consensus) |
| Refer persons with proteinuria (> 300 mg per 24 hours) to a nephrologist for further evaluation. (SOR: strong, based on low-quality evidence) |
| Screening for vascular disease and stroke risk |
| Screen annually with TCD according to methods used in the STOP studies,* beginning at 2 years of age and continuing until at least 16 years of age. (SOR: strong, based on moderate-quality evidence) |
| Refer children with conditional (170 to 199 cm per second) or elevated (≥ 200 cm per second) TCD findings to a subspecialist with expertise in long-term transfusion therapy aimed at preventing stroke. (SOR: strong, based on high-quality evidence) |
| Do not perform screening with CT or MRI in asymptomatic children with SCD. (SOR: moderate, based on low-quality evidence) |
| Do not perform screening with neuroimaging (TCD, CT, or MRI) in asymptomatic adults with SCD. (SOR: moderate, based on very low-quality evidence) |
| Do not perform screening with TCD in children with SCD but not SCA (e.g., HbSβ+-thalassemia, HbSC). (SOR: strong, based on low-quality evidence) |
In women with SCD, regular use of contraception can decrease the health risks associated with an unintended pregnancy. Progestin-only contraceptives (pills, injections, and implants) and barrier methods have no restrictions or concerns for use in women with SCD.17,18 The levonorgestrel-releasing intrauterine system is also associated with few risks, except for those associated with the device placement.17,18 Pregnancy in women with SCD is associated with preterm delivery, preeclampsia, stillbirth, high maternal mortality, and severe fetal anemia. Extra prenatal surveillance is recommended in consultation with a maternal-fetal subspecialist whenever possible.19,20 Both men and women with SCD require basic contraceptive information and referral for genetic counseling when making reproductive decisions.21
COMMON ACUTE AND CHRONIC COMPLICATIONS
Persons with SCD can have acute complications that require rapid interventions to avert or lessen the risk of life-threatening consequences. The most common complication is a vasoocclusive crisis (VOC). These episodes usually present with sudden onset of severe pain, often localized to the extremities, chest, or back, but with few objective findings. Nearly all persons with SCD will have a VOC during their lifetime,22,23 sometimes as early as six months of age, when it presents as dactylitis. Recurrences have variable presentations and frequency.23,24 VOC is a clinical diagnosis with no objective diagnostic tests. Other potential causes for the acute pain should be excluded while providing required analgesia. Primary management of VOC includes rapid triage, assessment, and administration of appropriate analgesics. Pain management should be based on patient-reported pain severity. For mild or moderate pain, treatment may begin with nonsteroidal anti-inflammatory drugs, if they are not contraindicated. For more severe pain, opioids are the first-line therapy; they should be provided immediately and therapy should be guided by the patient's history of previous successful regimens.25 Meperidine (Demerol) should not be used unless it is the only effective alternative available.26 Hydration and other nonpharmacologic therapies (e.g., maintaining body temperature) may also be useful.
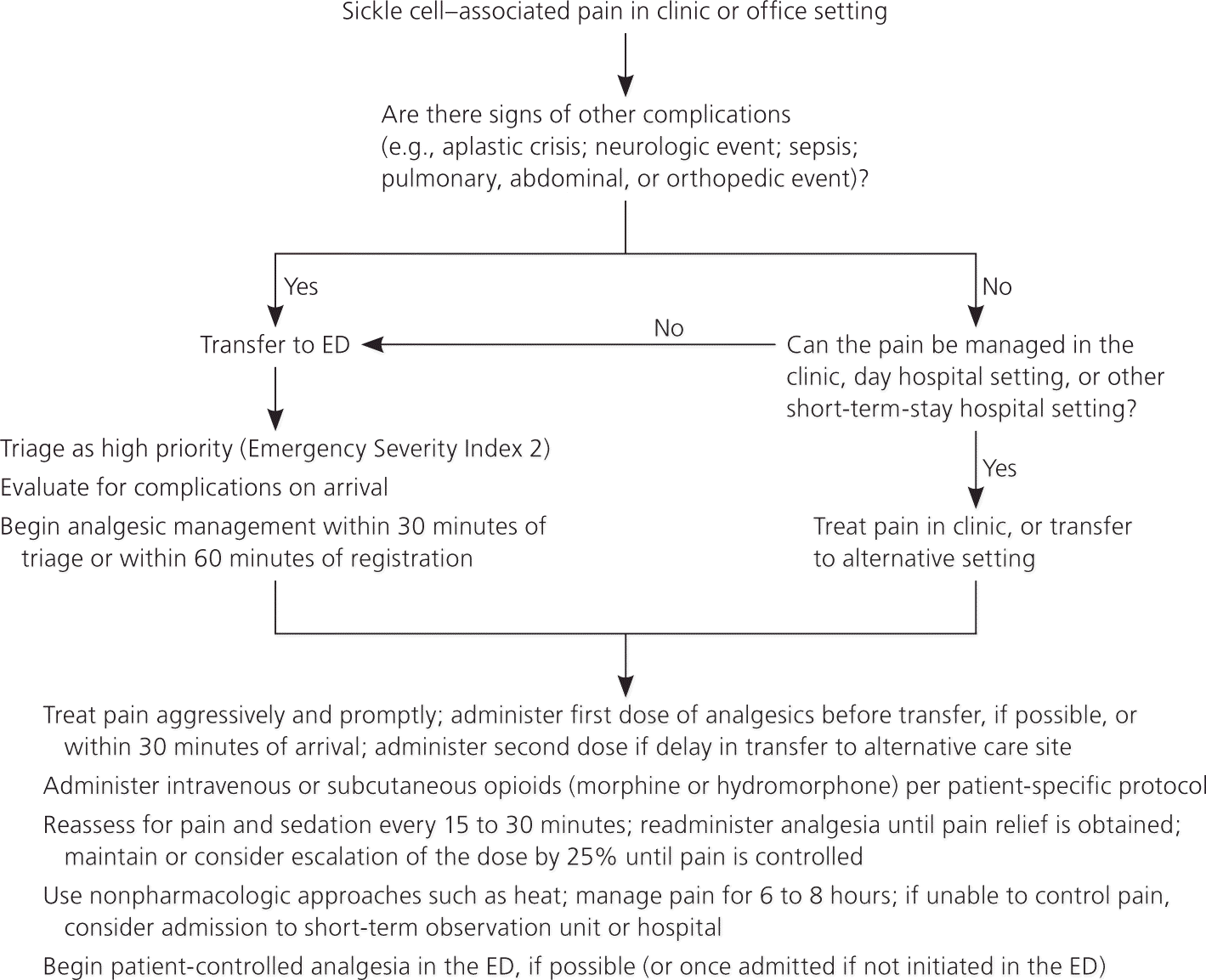
Concerns about drug-seeking behavior are pervasive and often triggered by patients with SCD who have frequent VOCs that require potent analgesia. These concerns should be addressed after adequately treating the acute pain. The use of an individualized VOC therapy plan that is carried with the patient and presented at urgent, emergency, or other care sites may lessen these concerns and facilitate rapid VOC management (Figure 1).1 Evaluation of suspected or possible drug-seeking behavior requires careful review of previous drug use, discussions with the patient's prescribing physicians, and objective information about drug abuse behavior. Consultation with a behavioral health expert is indicated when there are concerns about pain medication abuse.27
Acute chest syndrome (ACS), defined as the presence of a new lung infiltrate in a patient with acute onset of lower respiratory symptoms such as cough and shortness of breath, is less common than VOC but potentially life threatening. In children, it often presents with fever and signs of middle lobe lung involvement, whereas adults are often afebrile and have multilobe infiltrates. ACS can present on its own or as a complication of a VOC; it requires prompt evaluation and, once diagnosed, early intervention with antibiotic therapy and hospitalization.28,29 During hospitalization for a VOC, incentive spirometry can reduce the risk of ACS.30 When evaluating a new lung infiltrate in a patient with SCD, physicians should consider ACS before assuming the infiltrate represents community-acquired pneumonia.
Fever greater than 101°F (38.3°C), even in the absence of other signs of infection, should be evaluated carefully. Because of absent or reduced splenic function, persons with SCD have a high risk of overwhelming bacterial infections or sepsis.31 Fevers with possible infections should be treated empirically until culture results are available (Table 2).1

| ACS |
| Treat persons with SCD who have ACS with an intravenous cephalosporin, an oral macrolide antibiotic, and supplemental oxygen (to maintain oxygen saturation > 95%), and monitor for bronchospasm, acute anemia, and hypoxemia. (SOR: strong, based on low-quality evidence) |
| Encourage use of incentive spirometry while awake. (SOR: strong, based on moderate-quality evidence) |
| Consult an SCD expert regarding transfusion in persons with HbSC or HbSβ+-thalassemia who have ACS. (SOR: strong, based on low-quality evidence) |
| Perform urgent exchange transfusion in consultation with hematology, critical care, and/or apheresis subspecialists when there is rapid progression of ACS as manifested by oxygen saturation below 90% despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates, and/or decline in hemoglobin concentration despite simple transfusion. (SOR: strong, based on low-quality evidence) |
| Give simple blood transfusion (10 mL of red blood cells per kg) to improve oxygen-carrying capacity in persons with sickle cell anemia who have symptomatic ACS and whose hemoglobin concentration is > 1.0 g per dL (10 g per L) below baseline. If baseline hemoglobin is ≥ 9.0 g per dL (90 g per L), simple blood transfusion may not be required. (SOR: weak, based on low-quality evidence) |
| Priapism |
| Administer aggressive oral or intravenous hydration and oral or intravenous analgesia for episodes of priapism lasting 4 hours or longer. (SOR: strong, based on low-quality evidence) |
| Consult with a urologist for an episode of priapism lasting 4 hours or longer. (SOR: consensus) |
| Consult with a hematologist for possible preoperative transfusion if surgical intervention is required. (SOR: consensus) |
| Do not use transfusion therapy for immediate treatment of priapism associated with SCD. (SOR: moderate, based on low-quality evidence) |
| VOC |
| Initiate rapid treatment with parenteral opioids in adults and children with a VOC associated with severe pain. (SOR: strong, based on high-quality evidence) |
| Continue treatment with NSAIDs for adults and children with SCD who have a VOC associated with mild to moderate pain and who report relief with NSAIDs and do not have contraindications. (SOR: moderate, based on low-quality evidence) |
| Initiate around-the-clock opioid administration via patient-controlled analgesia or frequently scheduled doses vs. as-needed administration in adults and children with a VOC associated with severe pain. (SOR: moderate, based on low-quality evidence) |
| Use an individualized prescribing and monitoring protocol (written by the patient's primary physician for SCD care) or an SCD-specific protocol whenever possible to promote rapid, effective, and safe analgesic management and resolution of the VOC in children and adults with SCD (Figure 1). (SOR: consensus) |
| To reduce the risk of ACS, encourage the use of incentive spirometry while awake in adults and children who are hospitalized for a VOC. (SOR: strong, based on moderate-quality evidence) |
| Do not give blood transfusions to children and adults with a VOC unless there are other indications. (SOR: moderate, based on low-quality evidence) |
Hepatobiliary tract complications (e.g., cholelithiasis, acute cholecystitis, biliary sludge, acute choledocholithiasis) are common in persons with SCD, particularly in those with SCA. Recommended clinical responses are similar to those in patients without SCD who have these conditions. Gallstones occur in up to 75% of adults with SCD and, when asymptomatic, should be managed with watchful waiting.27
Disease-Modifying Therapies
Hydroxyurea therapy decreases SCD-related complications but is currently underused. Family physicians can be key to increasing the use of this therapy; they should ensure that every person affected by SCD is informed of the potential benefits and risks of hydroxyurea therapy, and should learn to initiate and monitor it.
Hydroxyurea works primarily by increasing the level of fetal hemoglobin (HbF), which does not sickle. This improves several clinical outcomes, such as decreasing the frequency of VOCs and ACS, reducing mortality, and decreasing the need for RBC transfusions and hospitalizations.32–34 Hydroxyurea is rapidly absorbed, has nearly complete bioavailability, and requires only once-daily oral dosing.
All persons nine months and older who have SCA are candidates for hydroxyurea therapy, although those with compound heterozygous conditions (e.g., HbSC, HbSβ+-thalassemia) require therapy less often than those with HbSS. Although the seminal randomized controlled trial of hydroxyurea therapy enrolled only adults with a history of moderate to severe VOCs, results from observational studies prompted the Expert Panel to broaden the recommendations to include adults with frequent VOCs or symptomatic anemia (Table 3).1 More recent studies included infants and young children without a history of VOCs, resulting in the recommendation to offer the therapy to even asymptomatic infants with SCA beginning at nine months of age.35–37 eTable A presents a consensus plan for initiating and monitoring hydroxyurea therapy.

| Beginning at 9 months of age, counsel all patients with SCA and their families about hydroxyurea therapy. (SOR: consensus) |
| Initiate hydroxyurea therapy in adults with SCA who have 3 or more sickle cell–associated moderate to severe pain crises in 12 months. (SOR: strong, based on high-quality evidence) |
| Initiate hydroxyurea therapy in adults with SCA who have a history of severe or recurrent acute chest syndrome. (SOR: strong, based on moderate-quality evidence) |
| Initiate hydroxyurea therapy in adults with SCA who have severe symptomatic chronic anemia that interferes with daily activities or quality of life. (SOR: strong, based on moderate-quality evidence) |
| Initiate hydroxyurea therapy in adults with SCA who have sickle cell–associated pain that interferes with daily activities and quality of life. (SOR: strong, based on moderate-quality evidence) |
| Offer hydroxyurea therapy for children 9 months and older who have SCA, regardless of clinical severity, to reduce SCD-related complications (e.g., pain, dactylitis, acute chest syndrome, anemia). (SOR for children 9 to 42 months of age: strong, based on high-quality evidence; SOR for children older than 42 months: moderate, based on moderate-quality evidence) |
| Use an established prescribing and monitoring protocol to ensure proper use of hydroxyurea therapy and to maximize its benefits. (SOR: strong, based on high-quality evidence) |
| Consider adding hydroxyurea therapy to improve anemia in adults and children with SCD and chronic kidney disease who are receiving erythropoietin. (SOR: weak, based on low-quality evidence) |
| Consult a sickle cell expert for consideration of hydroxyurea therapy in patients with HbSβ+-thalassemia or HbSC who have recurrent sickle cell–associated pain that interferes with daily activities or quality of life. (SOR: moderate, based on low-quality evidence) |
| Consult a sickle cell expert if a patient does not have a clinical response to appropriate doses and duration of hydroxyurea therapy. (SOR: moderate, based on very low-quality evidence) |
| Discontinue hydroxyurea therapy in pregnant or breastfeeding women. (SOR: moderate, based on very low-quality evidence) |

| Laboratory tests (recommended before starting therapy) | |
| CBC with WBC differential, reticulocyte count, platelet count, and RBC MCV | |
| Comprehensive metabolic profile, including kidney and liver function tests | |
| Pregnancy test for women | |
| Quantitative measurement of HbF, if available (e.g., hemoglobin electrophoresis, high-performance liquid chromatography) | |
| Initiating and monitoring therapy | |
| Baseline elevation of HbF should not affect the decision to initiate therapy | |
| Males and females of reproductive age should be counseled about the need for contraception while taking hydroxyurea | |
| Starting dosage for adults: 15 mg per kg per day (round up to the nearest 500 mg); 5 to 10 mg per kg per day in patients with chronic kidney disease | |
| Starting dosage for infants and children: 20 mg per kg per day | |
| Monitor CBC with WBC differential and reticulocyte count at least every 4 weeks when adjusting dosage | |
| Aim for a target absolute neutrophil count ≥ 2,000 per μL (2.0 × 109 per L); younger patients with lower baseline counts may safely tolerate absolute neutrophil counts as low as 1,205 per μL (1.2 × 109 per L) | |
| Maintain platelet count ≥ 80,000 per μL (80.0 × 109 per L) | |
| If neutropenia or thrombocytopenia occurs: | |
| Maintain hydroxyurea dosing | |
| Monitor CBC with WBC differential weekly | |
| When blood counts have recovered, reinstitute hydroxyurea at a dosage of 5 mg per kg per day lower than the dosage given before onset of cytopenia | |
| If dose escalation is warranted based on clinical and laboratory findings: | |
| Increase in increments of 5 mg per kg per day every 8 weeks | |
| Give until mild myelosuppression is achieved (absolute neutrophil count 2,000 to 4,000 per μL [2.0 × 109 to 4.0 × 109 per L]), up to a maximum of 35 mg per kg per day | |
| Once a stable dosage is established, laboratory monitoring should include CBC with WBC differential, reticulocyte count, and platelet count every 2 to 3 months | |
| Patients should be reminded that the effectiveness of hydroxyurea depends on their adherence to daily dosing, and should be counseled not to double doses if a dose is missed | |
| A clinical response to treatment may take 3 to 6 months; therefore, a 6-month trial at the maximum tolerated dose is required before considering discontinuation because of lack of adherence or nonresponse | |
| Monitor RBC, MCV, and HbF levels for evidence of consistent or progressive response | |
| A lack of increase in MCV or HbF is not an indication to discontinue therapy | |
| Long-term hydroxyurea therapy is indicated in patients who have a clinical response | |
| Therapy should be continued during hospitalizations or illness | |
Longer-term observational studies suggest that hydroxyurea therapy has long-term beneficial effects across all age groups with limited adverse effects—usually myelotoxicity in the form of reversible cytopenias, but no deleterious effects on growth and development. Thus far, there is no evidence of increased carcinogenicity or mutagenesis during long-term therapy in adults.38 However, hydroxyurea therapy is contraindicated in pregnant and breastfeeding women.27
Donor RBC transfusion, the first disease-modifying therapy used for SCD, reduces the percentage of circulating RBCs with HbS, thereby treating acute symptomatic anemia as well as treating and preventing many SCD complications. However, the need for repeated venous access and the associated risks and complications, such as alloimmunization and iron overload, limit its use.39,40
Donor RBCs may be administered as a simple transfusion—without removal of any recipient blood—or as an exchange transfusion, which involves removal of recipient blood before and possibly during infusion. Transfusions can be episodic or long term. Episodic transfusion is used acutely in response to an SCD complication or prophylactically in preparation for general anesthesia and surgery. Long-term transfusion therapy is used when sustained reduction of HbS (e.g., to less than 30%) is desired for primary or secondary prophylaxis for specific SCD complications, most commonly stroke in children.14–16

| Indications for acute transfusion | Type of transfusion |
|---|---|
| Symptomatic ACS and decreased hemoglobin of 1.0 g per dL (10 g per L) below baseline (SOR: weak, based on low-quality evidence) | Simple |
| Symptomatic severe ACS (oxygen saturation < 90% despite supplemental oxygen; SOR: strong, based on low-quality evidence) | Exchange |
| Acute splenic sequestration and severe anemia (SOR: strong, based on low-quality evidence) | Simple |
| Stroke (SOR: moderate, based on low-quality evidence) | Simple or exchange |
| Aplastic crisis (SOR: consensus) | Simple |
| Hepatic sequestration (SOR: consensus) | Simple or exchange |
| Intrahepatic cholestasis (SOR: consensus) | Simple or exchange |
| Multisystem organ failure (SOR: consensus) | Simple or exchange |
| Symptomatic anemia (SOR: consensus) | Simple |
| Contraindications | |
| Asymptomatic anemia (SOR: consensus) | |
| Priapism (SOR: moderate, based on low-quality evidence) | |
| Uncomplicated painful crisis (SOR: moderate, based on low-quality evidence) | |
Chronic blood transfusion programs are usually based in comprehensive SCD centers, but benefit from linkages to physicians who provide community continuity of care. Within the center, careful blood matching, including RBC phenotype matching to at least the C, E, and K antigens, decreases the incidence of alloimmunization and transfusion reactions. Detailed monitoring strategies must be used and continued into community practices to identify delayed transfusion reactions, which can present as acute anemia, pain, or jaundice days to weeks later; to maintain low HbS levels; and to track and treat transfusion-related iron overload.40 The Expert Panel supports quarterly monitoring of serum ferritin levels. Chelation therapy can effectively remove excess iron in patients with confirmed iron overload.27
Resources for Consultation
Family physicians may have limited experience in offering continuing or emergency care to patients with SCD, which makes it important to learn about consultative resources available regionally or nationally. Comanagement with adult or pediatric hematology-oncology subspecialists can enhance the family physician's knowledge and overall care of patients with SCD. In special cases (e.g., stroke, priapism, ACS), assistance can be garnered from other local subspecialists. However, local or regional subspecialists may also have limited experience in the care of SCD-related complications. Care of patients with SCD usually requires the involvement of a large group of health care professionals.
Data Sources: This is a summary of and highlights from evidence-based guidelines. We did not do any further searches than those completed for the guidelines. The methods for guideline development are described within the full guideline text (http://www.nhlbi.nih.gov/health-pro/guidelines/sickle-cell-disease-guidelines/index.htm). In summary, key questions (n > 40) were developed to guide selection of keywords used to search the available English-language literature from the Cochrane Database of Systematic Reviews, Bandolier, and the National Guideline Clearinghouse, and recommendations posted by the U.S. Preventive Services Task Force and the Advisory Committee on Immunization Practices.
The Evidence-Based Management of Sickle Cell Disease, Expert Panel Report 2014, is based on the best available but limited evidence. When high-quality evidence was lacking, expert consensus was used to provide recommendations. The guideline development methodology is described in detail in the full report.1
Some groups have used the guidelines to develop implementation strategies, which are presented as information without review or endorsement by the Expert Panel.
Suggestions from Community Care of North Carolina: https://www.communitycarenc.org/chacc-hematology-guidelines/
Summary of SCD guidelines from the National Heart, Lung, and Blood Institute (Quick Guide): http://www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/quickguide.pdf
The views expressed in this article are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute or the U.S. Department of Health and Human Services.
The authors thank George R. Buchanan, MD, and Lanetta Jordan, MD, MPH, MSPH, for assistance with the manuscript. They also thank the following panel members: George R. Buchanan, MD; Araba N. Afenyi-Annan, MD, MPH; Samir K. Ballas, MD; Kathryn L. Hassell, MD; Andra H. James, MD, MPH; Sophie M. Lanzkron, MD, MHS; Richard Lottenberg, MD; William J. Savage, MD, PhD; Paula J. Tanabe, PhD, RN; Russell E. Ware, MD, PhD; M. Hassan Murad, MD, MPH; Jonathan Goldsmith, MD; Eduardo Ortiz, MD, MPH; and Robinson Fulwood, PhD, MSPH.
