The vaso-occlusive crisis, or sickle cell crisis, is a common painful complication of sickle cell disease in adolescents and adults. Acute episodes of severe pain (crises) are the primary reason that these patients seek medical care in hospital emergency departments. Frequently, however, the pain is incompletely treated. Despite advances in pain management, physicians are often reluctant to give patients adequate dosages of narcotic analgesics because of concerns about addiction, tolerance and side effects. It is important to recognize a pain crisis early, correct the inciting causes, control pain, maintain euvolemia and, when necessary, administer adequate hemoglobin to decrease the hemoglobin S level. The family physician and the hematologist must work together to treat acute pain episodes promptly and effectively, manage the long-term sequelae of chronic pain and prevent future vaso-occlusive crises.
The vaso-occlusive crisis, or sickle cell crisis, is initiated and sustained by interactions among sickle cells, endothelial cells and plasma constituents.1 Vaso-occlusion is responsible for a wide variety of clinical complications of sickle cell disease, including pain syndromes, stroke, leg ulcers, spontaneous abortion and renal insufficiency.
Acute pain in patients with sickle cell disease is caused by ischemic tissue injury resulting from the occlusion of microvascular beds by sickled erythrocytes during an acute crisis. Chronic pain occurs because of the destruction of bones, joints and visceral organs as a result of recurrent crises. The effect of unpredictable recurrences of acute crises on chronic pain creates a unique pain syndrome.2,3
Acute bone pain from microvascular occlusion is a common reason for emergency department visits and hospitalizations in patients with sickle cell disease.4 Obstruction of blood flow results in regional hypoxemia and acidosis, creating a recurrent pattern of further sickling, tissue injury and pain. The severe pain is believed to be caused by increased intra-medullary pressure, especially within the juxta-articular areas of long bones, secondary to an acute inflammatory response to vascular necrosis of the bone marrow by sickled eythrocytes.5 The pain may also occur because of involvement of the periosteum or periarticular soft tissue of the joints.
The approach to pain control must include measures to treat acute pain crises, prevent future vaso-occlusive crises and manage the long-term sequelae of chronic pain that can result from multiple recurrent bony infarctions.
Natural History of the Vaso-occlusive Crisis
Epidemiologic data indicate that 5.2 percent of patients with sickle cell disease have three to 10 episodes of severe pain every year.6 In most patients, a pain crisis resolves within five to seven days. A severe crisis may cause pain that persists for weeks to months.2
When a vaso-occlusive crisis lasts longer than seven days, it is important to search for other causes of bone pain, such as osteomyelitis, avascular necrosis and compression deformities. When a recurrent bone crisis lasts for weeks, an exchange transfusion may be required to abort the cycle.
The frequency, severity, location and duration of pain crises can vary considerably, even within a specific disease subtype.4,7 [ corrected] Patients with homozygous sickle cell and sickle cell–β°-thalassemia have a higher frequency of vaso-occlusive pain crises than patients with hemoglobin SC and sickle cell–β+-thalassemia genotype.6,7 Disease severity is thought to depend on a complex interaction of genetic, rheologic and hematologic factors, as well as microvascular and endothelial factors.8–10
The psychologic, behavioral and cultural profile of individual patients also influences their perception of pain and their ability to cope with the pain.11 The impact that vaso-occlusive crises can have on a patient's life depends on the frequency and duration of each episode, the regions of the body that are affected and the intensity of the pain.2
Pain Patterns in the Vaso-occlusive Crisis
A vaso-occlusive crisis most commonly involves the back, legs, knees, arms, chest and abdomen.4,5,12 The pain generally affects two or more sites. Bone pain tends to be bilateral and symmetric. Recurrent crises in an individual patient usually have the same distribution.4,5,13 It has been postulated that the symmetry of bone marrow necrosis can be accounted for by centrally mediated reflexes that direct blood away from the marrow in response to the cooler skin temperature.13 If a patient's pain has a different than usual pattern, other causes for the pain should be sought.
An acute abdominal pain crisis often resembles an intra-abdominal process such as cholecystitis or appendicitis. Diagnoses that may require surgery and suggest a process other than vaso-occlusive crisis include pain in the absence of a precipitating event, pain that differs from the pain experienced in previous vaso-occlusive crises, and lack of pain relief within 48 hours despite hydration and oxygen therapy.14
Evaluation
The work-up of the patient with a vaso-occlusive crisis should include a complete history, a physical examination, selected laboratory tests and a search for reversible conditions known to precipitate pain crises. The physician should look for clinical evidence of dehydration and infection. The extent of bone and soft tissue involvement should also be assessed.
Routine laboratory testing is unnecessary in patients with uncomplicated vaso-occlusive crises. If a patient has symptoms that are severe enough to warrant hospitalization, laboratory tests should include a complete blood count, reticulocyte count and urinalysis. If fever is present, a chest radiograph should be obtained, and urine, sputum and blood should be cultured for a possible source of infection. Fever is common in patients with an uncomplicated vaso-occlusive crisis and does not necessarily indicate the presence of an underlying infection.5
Precipitating Factors
Predicting when a vaso-occlusive crisis may occur can be difficult. A number of factors, however, including dehydration, infection and cold weather, are known to precipitate acute crises (Table 1).2,4,5 In many patients, the exact cause cannot be determined.2
TABLE 1. Factors That Can Cause Sickle Cell Crises
Patients need to be aware of the factors that can precipitate vaso-occlusive crises. These patients are particularly susceptible to dehydration because of a reduced ability to conserve water secondary to a defect in renal concentrating ability. They should be counseled to wear warm clothes in cold weather, drink adequate amounts of fluids in hot weather and avoid exercising to the point of fatigue and dehydration (Table 2).
TABLE 2. Measures for Preventing Pain Crises in Patients with Sickle Cell Disease
| Consuming adequate amounts of fluids to prevent dehydration (especially during febrile periods and hot weather) |
| Avoiding mountain climbing or air flights in an unpressurized cabin (noncommercial flights) above 10,000 feet |
| Avoiding exposure to extreme cold, exercising to exhaustion or using drugs that can lead to acidosis (e.g., acetazolamide [Diamox]) |
| Genetic screening and vocational counseling about working (e.g., roofing) or taking part in extreme physical activity (e.g., military training) in the heat |
| Avoiding hypoxemia in the perioperative period when general anesthesia is used or when a procedure involves hypertonic radiographic dyes |
Medical Management
Acute sickle cell crises are managed primarily with drug therapy. Psychologic supportive care is also important. The standard treatment approach includes opioid analgesics, adequate hydration, rest, and cognitive and behavioral therapies.4
Optimal management requires a multidisciplinary team that includes a family physician, a hematologist, nurses, a psychiatrist, a physical therapist, a pain specialist and social workers. These team members work together to provide empathetic, consistent, longitudinal care in a trustworthy environment.2,4 Discussions of coping mechanisms, reassurance about pain management and the presence of a cohesive family unit are all important in preventing psychologic instability and the development of a chronic pain syndrome.
EVALUATION OF PAIN
Pain scales can be useful for quantifying the intensity of pain. These tools include the visual analog scale, the verbal categorical scale and the pain relief scale. Several of these measures are shown in Figure 1.4
FIGURE 1. Measures of Pain Intensity and Location
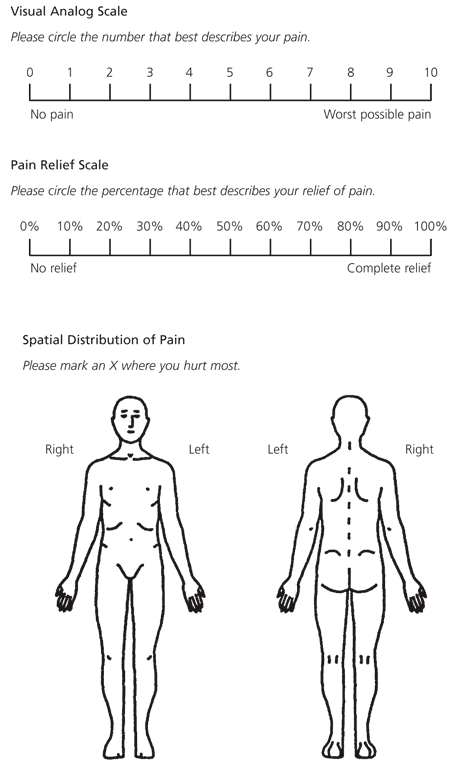
Techniques for determining the severity of pain and its location. (Top) The visual analog scale quantifies the intensity of pain. This scale is useful for titrating the dosages of narcotic analgesics and planning for hospital discharge. (Center) The pain relief scale compares the degree of pain relief that has been achieved with the degree of pain that the patient had on the previous day and/or the first day of hospitalization. (Bottom) The drawings are used to evaluate the spatial distribution of pain.
Adapted with permission from Ballis SK, Carlos TM, Dampier C, and Guidelines Committee. Guidelines for standard of care of acute painful episodes in patients with sickle cell disease. Harrisburg, Pa.: Commonwealth of Pennsylvania Department of Health, 1996.
The visual analog scale consists of a horizontal line labeled from zero to 10, with zero indicating “no pain” and 10 indicating “worst pain possible.”4 The patient circles the number that indicates the overall intensity of the pain. The visual analog scale has been found to be a clinically useful objective parameter for titrating narcotic analgesics and planning hospital discharge.12
The verbal pain scale can be used if a patient is unable to provide a written response to the visual analog scale. The patient is asked to report pain intensity verbally on a scale of zero (no pain) to 10 (worst pain possible).
The pain relief scale compares the degree of pain relief that has been achieved with the degree of pain the patient had on the previous day and/or the first day of hospitalization. The degree of pain relief is based on a scale of zero to 100 percent. 4
The patient can also be asked to mark the area of pain on a scaled body drawing (Figure 1).4 This technique is useful for determining the extent of involvement and for distinguishing the pain of vaso-occlusive crisis from pain caused by other complications, such as joint infection.12
SPECIFIC PAIN MANAGEMENT
Pain from a vaso-occlusive crisis is often undertreated because of concerns about narcotic addiction and tolerance, perceived drug-seeking behavior, excessive sedation, respiratory depression and lack of specific findings on the physical examination.2–4
Physicians often fail to prescribe narcotics appropriately and tend to overestimate opioid dependence in patients with pain crises. Yet the incidence of opioid analgesic addiction in patients with sickle cell disease has been reported to be no higher than 3 percent.15–17
Many drug regimens have been effective in the treatment of acute pain in sickle cell disease. Pain management should follow the three-step “analgesic ladder” recommended by the World Health Organization for the treatment of cancer-related pain.18 The choice of analgesic and the dosage used should be based on the severity of pain in the individual patient.
Patients with mild pain can often be treated at home with oral fluids and nonnarcotic analgesics4,19 (Table 3).3,4 Patients can also be started on acetaminophen with or without codeine or oxycodone (Roxicodone), depending on pain severity. Nonsteroidal anti-inflammatory drugs can be used unless they are specifically contraindicated because of peptic ulcer disease, renal disease or hepatic dysfunction.
TABLE 3. Nonnarcotic Analgesics for Mild Pain in Sickle Cell Disease
| Drug | Usual starting dosage in adults |
|---|---|
| Acetaminophen (Tylenol) | 500 to 1,000 mg every 4 to 6 hours (maximum < 4,000 mg per day) |
| Acetylsalicylic acid (aspirin) | 650 to 1,000 mg every 4 to 6 hours (maximum < 4,000 mg per day) |
| Diflunisal (Dolobid) | 1,000 mg initially, then 500 mg every 8 to 12 hours |
| Choline magnesium trisalicylate (Trilisate) | 1,000 to 1,500 mg every 12 hours |
| Ibuprofen (Advil) | 200 to 400 mg every 4 to 6 hours |
| Naproxen (Naprosyn) | 500 mg initially, then 250 mg every 6 to 8 hours |
| Fenoprofen (Nalfon) | 200 mg every 4 to 6 hours |
| Ketoprofen (Orudis) | 25 to 75 mg, then 250 mg every 6 to 8 hours (maximum < 300 mg per day) |
Narcotic analgesics can be used in patients with moderate to severe pain. The dosage of the selected narcotic should be titrated to achieve effective pain control.2,4,19 Because of the dose-limiting side effects of weak opioids (codeine and oxycodone), which include sedation, nausea and vomiting, these drugs are best used to manage moderate pain (Table 4).4 Pain that is sufficiently severe to require an emergency department visit or hospitalization should be treated with stronger opioids1,2,4,19,20 (Table 5).3,4,19
TABLE 5. Opioids Used to Treat Severe Pain in Sickle Cell Disease
| Drug | Oral/IM potency | Equianalgesic dosages | Usual starting dosage in adults | ||
|---|---|---|---|---|---|
| IM | Oral | Oral | Parenteral | ||
| Morphine (Duramorph) | 6* | 10 mg | 60 mg | 15 to 30 mg every 4 hours | 0.1 to 0.15 mg per kg every 3 or 4 hours |
| Hydromorphone (Dilaudid-Hp) | 5 | 1.5 mg | 7.5 mg | 2 to 4 mg every 4 to 6 hours | 1 to 2 mg every 4 to 6 hours |
| Meperidine (Demerol) | 4 | 75 mg | 300 mg | 50 to 150 mg every 3 or 4 hours | 75 to 100 mg every 3 or 4 hours |
| Levorphanol (Levo-Dromoran) | 2 | 2 mg | 4 mg | 2 to 4 mg every 6 to 8 hours | Up to 1 mg IV every 3 to 6 hours; 1 to 2 mg IM or SC every 6 to 8 hours |
IM = intramuscular; IV = intravenous; SC = subcutaneous.
*—Single-dose studies determined that the relative potency is 6:1; with repetitive doses, this ratio changes to 3:1.
If a patient has poor venous access and is unable to take enteral narcotics because of vomiting, the subcutaneous route can be employed, using morphine or its equivalent. It is important to remember that subcutaneous administration may result in prolonged absorption if a patient is dehydrated.
Adjuvant nonopioid agents such as antihistamines and antiemetics can be helpful for preventing or relieving opioid-related side effects.4,19 The use of adjuvant analgesics such as tricyclic antidepressants should be considered in patients with a chronic pain syndrome resulting from recurrent acute pain crises.21
Nonpharmacologic techniques can also be tried. These measures include physical therapy, rest, heat application, transcutaneous electrical nerve stimulation (TENS), self-hypnosis and diversional techniques.2,4,19,22
MEPERIDINE VS. MORPHINE
In the past, moderate to severe pain in sickle cell disease was usually treated with meperidine (Demerol) administered parenterally or, more commonly, intramuscularly. Compared with morphine, however, meperidine has a number of properties that make it a poor opioid analgesic for repeated use in most patients with sickle cell disease.
Meperidine is a weak opioid analgesic with a short half-life (two to three hours). Thus, frequent dosing is required to maintain a sustained analgesic effect. In addition, normeperidine, a metabolite of meperidine, has been associated with seizures, particularly in patients with impaired renal function who are being given high doses at frequent intervals.3,4 Repeated injections of meperidine can lead to fibrosis, infection and sterile abscess formation at the injection site.
For these reasons, parenterally administered morphine should be considered the treatment of choice for moderate to severe pain in vaso-occlusive crises. Morphine's side effects include pruritus, nausea, vomiting and rash. In addition, dosage adjustments are necessary in patients with liver dysfunction.19
Some patients prefer meperidine for the treatment of pain crises and may be reluctant to change to morphine. Analgesia should be discussed when patients are not in pain.
Regardless of the type of opioid analgesic used, respiratory rate and oxygen saturation must be closely monitored because of the potential for respiratory depression. If the respiratory rate is less than 10 per minute or excessive sedation occurs, the opiate should be discontinued, the dosage should be reduced or the dosing frequency should be lengthened.19,20
METHODS OF NARCOTIC DELIVERY
With parenteral administration, narcotic analgesics can be given using a fixed schedule (with rescue doses administered for breakthrough pain), continuous infusion or patient-controlled administration. Dosing on an “as-needed” basis should be avoided because it does not confer an adequate sustained level of analgesia.
Patient-controlled analgesia offers several unique advantages in the treatment of severe pain occurring in a vaso-occlusive crisis.15,23,24 One study24 found that the intermittent fixed schedule and the patient-controlled method were equally efficacious in providing adequate analgesia. Patient-controlled analgesia prevents fluctuation in blood drug levels and may reduce the time between the perception of pain and the administration of the analgesic. This approach reduces overmedication and excessive sedation. It also provides patient autonomy and decreases the nursing time required for analgesic administration.
OXYGEN MANAGEMENT
Oxygen therapy is often used routinely in the management of vaso-occlusive crises, despite lack of evidence supporting the effectiveness of these measures in all patients.25,26 Oxygen therapy has not been shown to affect the duration of a pain crisis or to be useful in patients with acute chest syndrome whose partial pressure of arterial oxygen (PaO2) is in the normal range. Hence, oxygen should be administered only if hypoxemia is present.1,25,26 Oxygen may also suppress erythrocyte production, depress reticulocytosis and cause rebound sickle cell crises on discontinuation of therapy when the arterial oxygen tension is raised above the normal range.27
Pulse oximetry may not be a reliable method of determining the PaO2 in patients with sickle cell disease.28 One reason may be the differences in the oxygen dissociation curve between normal hemoglobin and sickle cell hemoglobin (hemoglobin S).28 Sickle cell erythrocytes have decreased oxygen affinity and increased unsaturated hemoglobin in the arterial blood. All low pulse-oximetry saturation values should be compared with values obtained at steady state, if available, or should be confirmed by measuring the PaO2 directly with an arterial blood gas determination.29
TRANSFUSION
Most patients with sickle cell anemia have hemoglobin values of 6 to 10 g per dL (60 to 100 g per L). The hemoglobin S molecule has a low affinity for oxygen (which allows for adequate tissue oxygenation). During a vaso-occlusive crisis, a patient's hemoglobin level often declines by at least 1 g per dL (10 g per L).
A hemoglobin value of 5 g per dL (50 g per L) or less or a decline in the hemoglobin value of greater than 2 g per dL (20 g per L) from the patient's baseline level has been used as a guide for considering simple transfusion therapy.30 Patients should be transfused to their baseline hemoglobin level. A higher hematocrit may make the blood more viscous and further increase sickling.
Other indications for transfusion include acute chest syndrome with hypoxia and the need for surgery using general anesthesia.
Exchange transfusions should be considered only in patients who have a prolonged refractory vaso-occlusive crisis with a stable baseline hemoglobin concentration. The goal is to reduce sickling by reducing the hemoglobin S level to below 20 percent.30
FLUID REPLACEMENT
Increased plasma osmolarity from a reduced plasma volume can worsen a vaso-occlusive crisis by causing intracellular dehydration, hemoglobin polymerization and further sickling. During hyponatremia, the affinity of hemoglobin S for oxygen is increased. Therefore, at any given PaO2, less oxygen is in the deoxygenated state, which is the form most susceptible to polymerization. Patients with sickle cell disease have isosthenuria, which leads to difficulty in excreting a sodium load.28
Fluids should be administered in a quantity sufficient to correct existing deficits and replace ongoing losses in order to maintain a euvolemic state. Large fluid volumes may decrease plasma oncotic pressure and increase hydrostatic pressure. This can lead to pulmonary edema, especially in patients with impaired renal function, cardiac failure or pulmonary vascular injury.
If tolerated, oral rehydration should be used in patients with milder vaso-occlusive crises. The parenteral route of rehydration is indicated in patients with severe pain, vomiting or volume depletion. After existing volume deficits have been corrected with normal saline, fluid replacement should consist of 5 percent dextrose in water or in a 25 percent normal saline solution.28
HYDROXYUREA
Hydroxyurea (Hydrea) increases the production of hemoglobin F and thereby reduces the severity of sickle cell disease by preventing the formation of hemoglobin S polymers.1 At present, hydroxyurea should be used in patients who have severe complications and who can reliably follow the regimen.
Hydroxyurea is initiated in a dosage of 500 mg per day. The dosage is increased to 1,000 mg per day after six to eight weeks, with the patient monitored for a decline in granulocyte or platelet counts. The maintenance dosage is between 1,000 and 2,000 mg per day, depending on the balance between hematologic toxicity and increases in hemoglobin F values.1 Blood counts should be followed every four to six weeks to detect longer term hematologic toxicities.
A rising mean corpuscular volume is a good surrogate marker for rising hemoglobin F levels. Treatment should be stopped if a patient does not respond after several months of hydroxyurea therapy.
The long-term effects of hydroxyurea maintenance therapy are not well known. More studies are needed to better determine negative effects such as carcinogenicity and positive effects such as the prevention of organ damage and reduced mortality.
Management After Hospital Discharge
Most patients have residual pain at the time they are discharged from the hospital. Therefore, they should be given an oral narcotic analgesic in a dosage equivalent to the dosage that was necessary to control their pain while they were hospitalized. They should be given enough of the narcotic analgesic to last until the next scheduled outpatient follow-up visit.4,19
Patients should be instructed to use the visual analog scale or the verbal categorical scale as a guide for self-tapering of the analgesic dosage based on their level of pain.
The management of acute pain in sickle cell crises is summarized in Table 6.1–4,19 In addition, the American Pain Society recently released a comprehensive guideline on pain management in sickle cell disease.31
TABLE 6. Treatment Principles for Acute Pain Management in Patients with Sickle Cell Crises
| General principles |
| If possible, identify and treat underlying precipitating factors. |
| If needed, administer fluids orally, or intravenously as 5 percent dextrose in water or in a 25 percent normal saline solution. |
| Use oxygen therapy only if hypoxemia is present. |
| Acute pain management |
| Avoid delays in administering analgesia. |
| Administer an opioid analgesic parenterally (preferably intravenously) on a regular basis in a full therapeutic dosage or by patient-controlled analgesia. Avoid “as-needed” dosing. |
| Reassess the patient every 30 minutes for pain severity, sedation, vital signs and respiratory rate. |
| Use pain measurement scales as an objective guide to titrate the maintenance dosage of an analgesic and to determine treatment effects. |
| For breakthrough pain, administer one fourth to one half of the maintenance dosage, depending on the degree of sedation. |
| If three or more rescue doses are needed within a 24-hour period, increase the maintenance dosage by 25 to 50 percent, and repeat the same steps until analgesia is achieved. |
| Pain management after an acute crisis |
| Begin tapering the parenterally administered analgesic when the pain severity score is less than 5 on the visual analog scale or verbal pain scale and the patient's mood improves. Reduce the maintenance dosage by 25 percent every 24 hours, and replace the parenterally administered drug with an equianalgesic oral agent given in divided doses. |
| Consider hospital discharge when the patient's pain is controlled with an orally administered analgesic or no analgesia is needed. |
| If the patient still has pain at the time of hospital discharge, provide a prescription for a sufficient quantify of analgesic drug to treat resolving or relapsing pain until the patient's next office appointment. |
