Prescribers seek to provide their patients with access to the latest innovations in medicine to maximize their health status. When a new drug comes to market, it often has not been as widely tested as other available therapies, and its effectiveness and safety cannot be fully evaluated. To address this problem, physicians can use the STEPS (Safety, Tolerability, Effectiveness, Price, and Simplicity) mnemonic to provide an analytic framework for making better decisions about a new drug's appropriate place in therapy. A key element is to base this evaluation on patient-oriented evidence rather than accept disease-oriented evidence (which may be misleading), while avoiding inappropriate reliance on studies that report only noninferiority results or relative risk reductions. The primary question to ask for each new drug prescribing decision is, “Is there good evidence that this new drug is likely to make my patient live longer or better compared with the available alternatives?”
The prescribing of new drugs remains a controversial subject. Some new drugs offer distinct advantages over current therapies, whereas many offer little worthwhile advantage to patient care and are sometimes viewed by the drug companies as a means to retain or increase their market share. This might include “me-too” drugs or drugs for mild or poorly defined conditions. Other pharmaceutical targets include persons who are otherwise healthy or those deemed “at risk,” in whom the potential market share is big and more easily developed.
SORT: KEY RECOMMENDATIONS FOR PRACTICE

| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Patient-oriented evidence relating to morbidity, mortality, or quality of life leads to more reliable prescribing decisions for new drugs than traditional disease-oriented evidence using surrogate markers, such as physiologic variables, test results, and risk assessments. | C | 8 |
| The demonstration that a marker is independently associated with risk does not mean that treatments that modify levels of the marker will also modify clinical risk. | C | 11 |
| Clinical significance of new drugs is more reliably assessed by evaluating absolute risk reduction and the number needed to treat than by evaluating only relative risk reduction data. | C | 16 |
| New formulations of existing drugs or newer versions of previously marketed drugs, including extended-release versions, pro-drugs, metabolites, and isomers, are not necessarily safe or more effective than older versions. | C | 17 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
When a new drug is launched, it has not been as widely tested as other available therapies, and there is often insufficient good-quality published evidence to be able to judge effectiveness. Furthermore, the safety profile cannot be fully assessed, because only a limited number of patients will have been exposed to the drug by the time of licensing. This presents a dilemma for innovative physicians who seek to provide their patients with the best available therapeutic intervention, without exposing them to unknown risks.
A careful and critical approach to the use of new drugs is needed to ensure their use is appropriate. Physicians must be vigilant to detect and report possible adverse effects. This article suggests an analytic framework that can be used to help with this dilemma.
Factors to Help Make Prescribing Decisions
The STEPS (Safety, Tolerability, Effectiveness, Price, and Simplicity) mnemonic has been widely used to balance a prescribing decision.
SAFETY
It has been estimated that approximately 6.5 percent of patient hospital admissions are because of adverse drug reactions.1 One study suggested that adverse drug reactions cause death in 0.1 percent of medical inpatients and 0.01 percent of surgical inpatients.2 They may also cause considerable morbidity. Safety problems of new drugs are not always detected during clinical trials used for licensing purposes and may take many years to emerge. A study of new drugs introduced to the United States between 1975 and 1999 found that 10 percent had serious adverse effects that emerged only after approval.3 Furthermore, from 1998 to 2004, U.S. postmarketing surveillance was only able to identify serious adverse drug reactions a median of three years after approval.4 Similarly, 40 percent of safety withdrawals in Canada from 1963 to 2004 were within three years of registration.5
There are many examples of drugs that became widely used only to be withdrawn from the market because the harms were considered to outweigh any benefits. Examples include thalidomide (Thalomid), because phocomelia occurred in infants born to women who took the drug in early pregnancy, and diethylstilbestrol (Stilphostrol), because in utero exposure caused the subsquent development of clear cell adenocarcinoma of the vagina in young women. A recent high-profile example of this is rofecoxib (Vioxx), which was withdrawn because of increased risk of serious thrombotic events (including myocardial infarction and stroke) following long-term use compared with placebo.
Such problems may occur because licensing decisions are based on short-term clinical trial evidence involving approximately 1,500 patients.6 Putting this in perspective, more than 30,000 patients would need to be studied to have some certainty of detecting an adverse event with an incidence of one in 10,000. However, a serious adverse drug reaction with an incidence of one in 5,000 may be enough to cause withdrawal of the drug.
TOLERABILITY
Drugs are commonly prescribed to reduce the risk of a future event rather than to treat symptoms (e.g., statins in the primary prevention of cardiovascular disease). However, if use of such a drug also makes the person feel sick, they may be unlikely to continue to take the drug as prescribed, which is something the patient may not want to share with the prescriber.
EFFECTIVENESS
It has been suggested that up to 97 percent of published drug studies focus only on short-term effectiveness in trials investigating surrogate end points.7,8 Such studies can provide only disease-oriented evidence (evidence based on surrogate markers of health [e.g., blood tests, blood pressure, peak flow, electrocardiography patterns]). More helpful would be patient-oriented evidence that addresses real-life questions of whether patients actually live longer or better (e.g., reduced hospitalization, reduced mortality, improved quality of life).
History reveals numerous examples of drugs incorporated into clinical practice based on assumptions made from trials using surrogate markers of disease, only to be shown later to be wrong and harmful. Milrinone, for example, was used to improve cardiac output and exercise tolerance in patients with cardiac failure, but was subsequently shown to increase mortality.9 The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) demonstrated that doxazosin (Cardura) increased the risk of heart failure compared with a thiazide diuretic in the treatment of hypertension.10
The demonstration that a marker is independently associated with risk does not mean that treatments that modify levels of the marker will also modify clinical risk.11 Menopausal hormone therapy, for example, was known to reduce total and low-density lipoprotein cholesterol. This was expected to translate into less cardiovascular disease and deaths in users. The publication of the Women's Health Initiative study, however, showed that this therapy actually increases the risk of cardiovascular disease compared with placebo.12
More recently, torcetrapib was developed to increase high-density lipoprotein cholesterol concentrations. It was expected to lead to beneficial outcomes, but instead increased the risk of cardiovascular events compared with placebo, and was never approved by the U.S. Food and Drug Administration.13
Increasingly, physicians are realizing that just because a drug reduces a clinical risk factor, this does not necessarily predict that it will provide a worthwhile benefit to patients in the real world. Physicians must always ask the question, “Is there good evidence that this new drug is likely to make my patient live longer or better compared with the available alternatives?”
PRICE
The cost of a new drug should also be considered in the overall assessment of its place in therapy. The following example is based on an argument made in an analysis of the incremental cost-benefits of new technologies.14
Consider two drugs: drug A is an established agent and costs $20 per month for a course of treatment and produces a cure rate of 40 percent (i.e., 40 out of 100 persons who take it are cured of a particular disease). Drug B is a new drug that costs $50 per month and cures 50 percent of persons with the disease.
First impressions suggest that drug B is the logical choice to be used for all patients, because it cures more patients. However, consider how best to spend $100,000 (for example) on a particular disease each month. If drug A is used, 5,000 patients can be treated within the budget, and 2,000 (40 percent) of them will be cured. If the more effective, but more expensive, drug B is used, then only 2,000 patients can be treated within the budget, and only 1,000 (50 percent) of them will be cured. A policy of using drug B, even though it is more effective, will result in fewer patients being cured for the same amount of resource. This is a relatively simplistic way of looking at this dilemma. In reality, it is often more complex than this; the relative merits of each drug will also depend on the disease being treated.
SIMPLICITY
It is thought that between one half and one third of all drugs prescribed for long-term conditions are not taken as recommended.15 Thus, making a drug regimen as simple as possible is an important step in achieving compliance. Although a common sense approach can be applied to this (e.g., avoiding prescribing a drug that requires a dosing schedule of four times a day when a once-daily regimen is available), there is little good-quality evidence that using a once-daily formulation or combination product will improve adherence. Although reducing the complexity of a regimen can increase adherence, the evidence from qualitative interviews indicates that the difficulty for patients is integrating the regimen into their lives rather than dose complexity per se.15
Going Beyond STEPS: Other Issues to Consider
ABSOLUTE vs. RELATIVE RISK REDUCTIONS
Advertising materials and results from clinical trials investigating new drugs commonly report relative risk reductions rather than absolute risk reductions; this may exaggerate the perceptions of benefit or harm and can be misleading 8 (Table 1). For example, one study evaluated 222 articles published in leading medical journals and found that 68 percent of the articles did not report absolute risk.16 The clinical significance of new drugs is more reliably assessed by evaluating absolute risk reduction and number needed to treat than by evaluating only relative risk reduction data.16
Table 1. The Importance of Absolute vs. Relative Risk Reduction in the Evaluation of New Drugs

| Reporting RRR rather than ARR may exaggerate the perceptions of benefit or harm, and can be misleading. |
| Consider drug A, which reduces mortality from 40 to 30 percent if taken for one year. In this case: |
|
| Now consider use of drug A by patients with a lower baseline risk of dying (e.g., the drug reduces mortality from 1.00 to 0.75 percent if taken for one year). In this case: |
|
| Patients with the highest baseline risk benefit the most from treatment, but all patients need to take the drug for the few to benefit. However, all patients taking the therapy risk the possibility of adverse effects, and the treatment must be funded for all. |
ARR = absolute risk reduction; NNT = number needed to treat; RRR = relative risk reduction.
DO NEW VERSIONS OF OLD DRUGS ADD VALUE?
In an attempt to shortcut drug development, there is an increasing trend among pharmaceutical companies to bring to market new formulations of existing drugs or newer versions of previously marketed drugs, including extended-release versions, pro-drugs, metabolites, and isomers (Table 2).17 Changes such as these add value if they can demonstrably be shown to improve safety, effectiveness, or adherence to treatment. However, the timing of the introduction of such changes in formulation has suggested that this is often little more than an attempt to extend patent life of the drug in the face of new competition from generic drug manufacturers.17
Table 2. Examples of New Drugs “Created” from Old Drugs
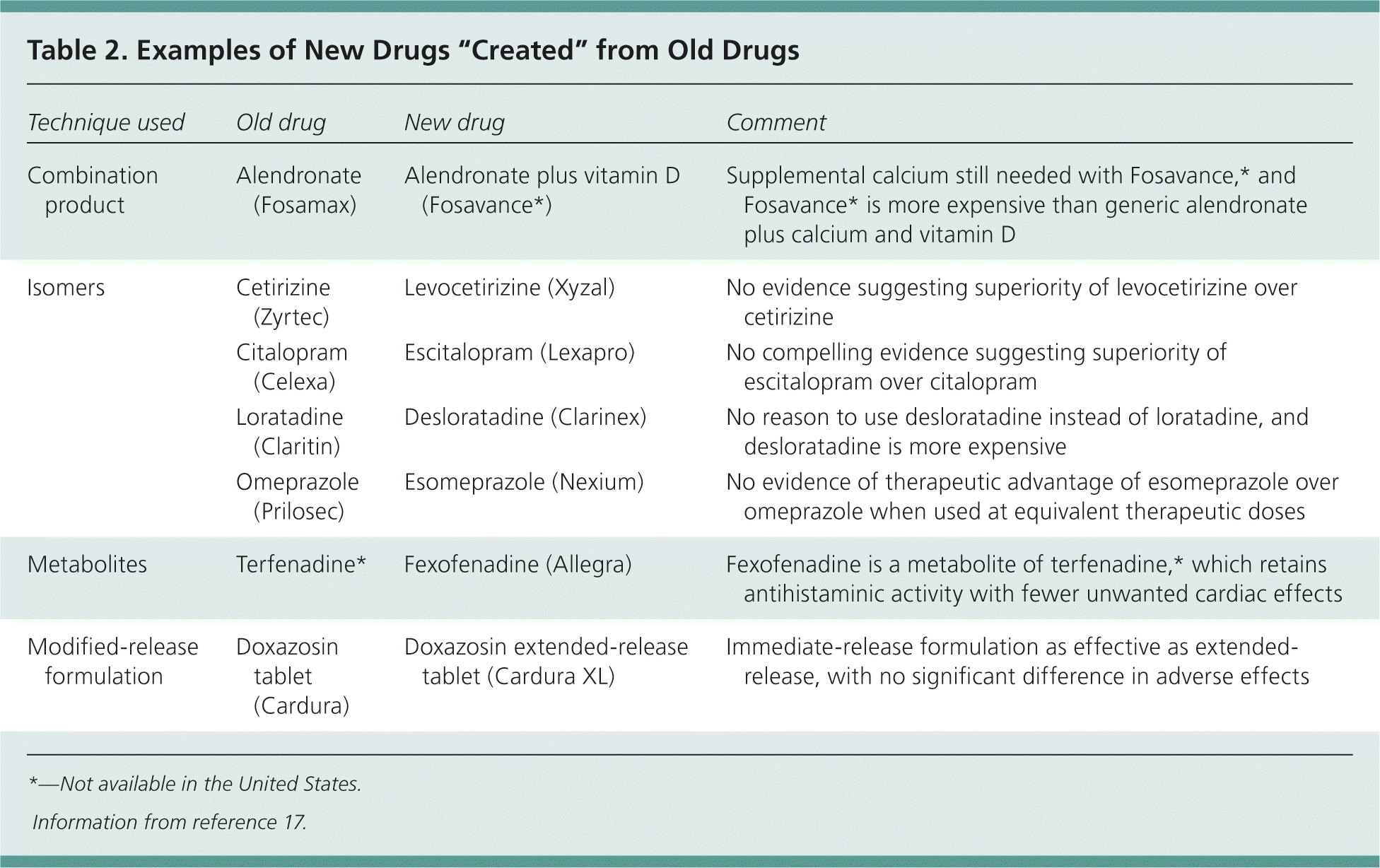
| Technique used | Old drug | New drug | Comment |
|---|---|---|---|
| Combination product | Alendronate (Fosamax) | Alendronate plus vitamin D (Fosavance*) | Supplemental calcium still needed with Fosavance,* and Fosavance* is more expensive than generic alendronate plus calcium and vitamin D |
| Isomers | Cetirizine (Zyrtec) | Levocetirizine (Xyzal) | No evidence suggesting superiority of levocetirizine over cetirizine |
| Citalopram (Celexa) | Escitalopram (Lexapro) | No compelling evidence suggesting superiority of escitalopram over citalopram | |
| Loratadine (Claritin) | Desloratadine (Clarinex) | No reason to use desloratadine instead of loratadine, and desloratadine is more expensive | |
| Omeprazole (Prilosec) | Esomeprazole (Nexium) | No evidence of therapeutic advantage of esomeprazole over omeprazole when used at equivalent therapeutic doses | |
| Metabolites | Terfenadine* | Fexofenadine (Allegra) | Fexofenadine is a metabolite of terfenadine,* which retains antihistaminic activity with fewer unwanted cardiac effects |
| Modified-release formulation | Doxazosin tablet (Cardura) | Doxazosin extended-release tablet (Cardura XL) | Immediate-release formulation as effective as extended-release, with no significant difference in adverse effects |
*— Not available in the United States.
Information from reference 17.
Combination products often claim to improve adherence to therapy by reducing the number of tablets that a patient is required to take. Although some combination preparations can be useful (for example, if the patient is already stabilized on the individual components), this may not always be the case. One example is Fosavance (not available in the United States), which consists of alendronate (Fosamax) plus vitamin D. Fosavance still requires patients to take supplemental calcium, as well as the combination product. As such, this regimen does not appear to simplify the daily drug routine for patients, yet costs more than generic alendronate plus calcium and vitamin D.
NONINFERIORITY TRIALS
New drugs are often licensed following submission of evidence derived from noninferiority trials to the regulatory authorities. Normally, a randomized controlled trial tests one drug against another to see which is superior. Noninferiority trials aim to assess whether an intervention (e.g., a new drug) is no worse than a comparator (usually “standard” therapy); they essentially test similarity within a limit. Whether such trials are ethical has been questioned,18 because some consider that noninferiority trials are undertaken with predominantly commercial aims to allow entry of the drug to the market, but without helping to address questions related to the drug's appropriate place in therapy. Instead, marketing subsequently seeks to highlight some putative added value or perceived benefit to patients of other factors associated with the drug, such as faster absorption, extended duration of action, or ease of use. More concerning is the possibility that the noninferior drug, while providing no clinical advantages, may actually be less effective or less safe than existing therapy, because of the limit of noninferiority within the trial being set too generously. This has major implications for drugs that are potentially toxic, such as those used in oncology, in which small differences in effectiveness and tolerability can have a big impact on the patient's quality of life and mortality.
Final Comments
Using the STEPS mnemonic and considering the previously mentioned issues will allow busy physicians to objectively focus their assessment of new drugs so they distinguish marketing claims from true effectiveness advantages. Physicians should avoid becoming preoccupied with a drug's mechanism of action and be aware that treatments that modify levels of a surrogate marker of disease will not necessarily modify clinical risk. There are many examples in which application of such disease-oriented evidence has been proven wrong and led to patient harm. Routinely asking, “Is there good evidence that this new drug is likely to make my patient live longer or better compared with the available alternatives?” will help ensure that patients consistently receive the safest and most cost-effective treatment.
