One in 2,000 children younger than 18 years is thought to have a primary immunodeficiency disease. Antibody, combined B-cell and T-cell, phagocytic, and complement disorders are the most common types. Children with these diseases tend to have bacterial or fungal infections with unusual organisms, or unusually severe and recurrent infections with common organisms. A family history of primary immunodeficiency disease is the strongest predictor of a person having this type of disease. When an immunodeficiency disease is suspected, initial laboratory screening should include a complete blood count with differential and measurement of serum immunoglobulin and complement levels. The presence of lymphocytopenia on complete blood count suggests a T-cell disorder, whereas a finding of neutropenia suggests a phagocytic disorder. Abnormal serum immunoglobulin levels suggest a B-cell disorder. Abnormalities on assay of the classic or alternative complement pathways suggest a complement disorder. If laboratory results are abnormal, or if clinical suspicion continues despite normal laboratory results, children should be referred for further evaluation. Human immunodeficiency virus infection should also be considered, and testing should be performed, if appropriate; this infection often clinically resembles a T-cell disorder.
More than 180 primary immunodeficiency diseases have been identified, and that number is growing as advances in genetic technology allow for further identification of specific defects of immunity.1–3 The prevalence of these diseases varies. In Sweden, for example, the prevalence is 0.35 per 100,000 persons, whereas in southern Australia, the prevalence is 12.4 per 100,000 persons.4 In the United States, the overall prevalence of diagnosed primary immunodeficiency disease is estimated to be one in 1,200 persons; among children younger than 18 years, it is estimated to be one in 2,000.5 The overall incidence in the United States is 10.3 per 100,000 person-years6; the median interval from onset of symptoms to diagnosis is 2.7 years.6
SORT: KEY RECOMMENDATIONS FOR PRACTICE

| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Physicians should suspect T-cell disorders and human immunodeficiency virus infection in children who have diarrhea, show failure to thrive, and have opportunistic infections. | C | 3 |
| Physicians should suspect a primary immunodeficiency disease in children who have unusually severe and recurrent infections with common pathogens, or infections with unusual pathogens. | C | 3 |
| When a primary immunodeficiency disease is suspected in a child, initial laboratory testing should include a human immunodeficiency virus test, complete blood count with differential, and measurement of serum immunoglobulin and complement levels. | C | 3, 7, 10, 14 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
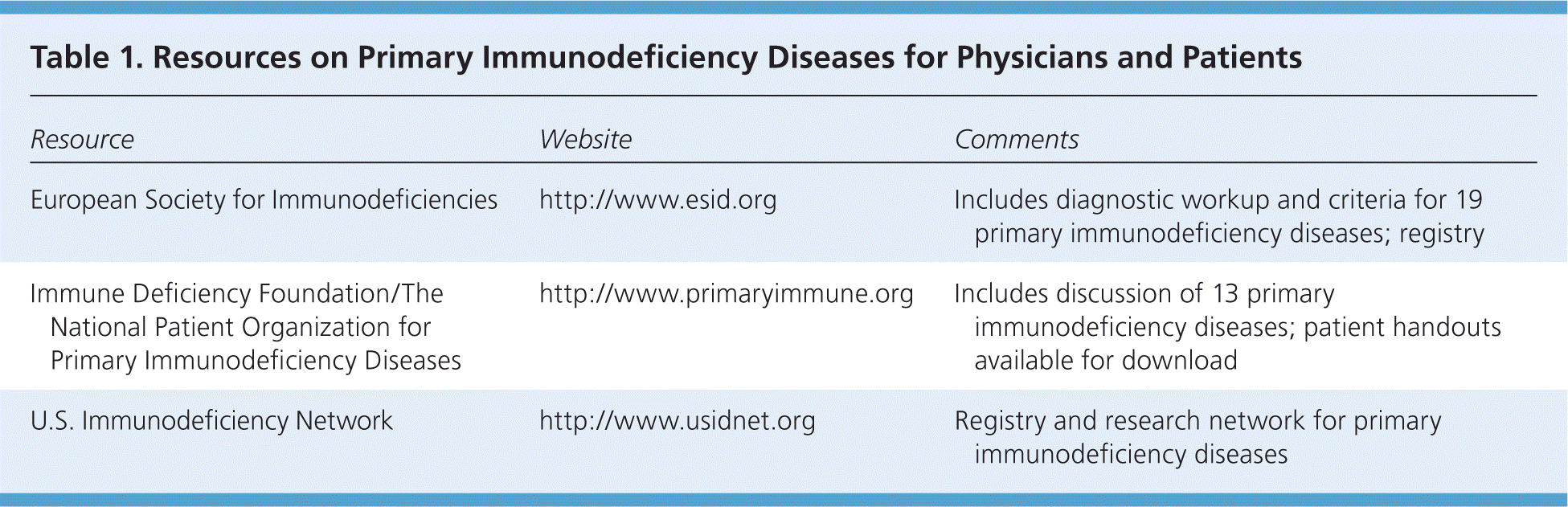
The European Society for Immunodeficiencies (ESID) has the largest registry of primary immunodeficiency diseases, with information on more than 16,000 patients and eight broad categories of disorders.4 Additional resources for physicians and patients are listed in Table 1.
Table 1. Resources on Primary Immunodeficiency Diseases for Physicians and Patients
| Resource | Website | Comments |
|---|---|---|
| European Society for Immunodeficiencies | http://www.esid.org | Includes diagnostic workup and criteria for 19 primary immunodeficiency diseases; registry |
| Immune Deficiency Foundation/The National Patient Organization for Primary Immunodeficiency Diseases | http://www.primaryimmune.org | Includes discussion of 13 primary immunodeficiency diseases; patient handouts available for download |
| U.S. Immunodeficiency Network | http://www.usidnet.org | Registry and research network for primary immunodeficiency diseases |
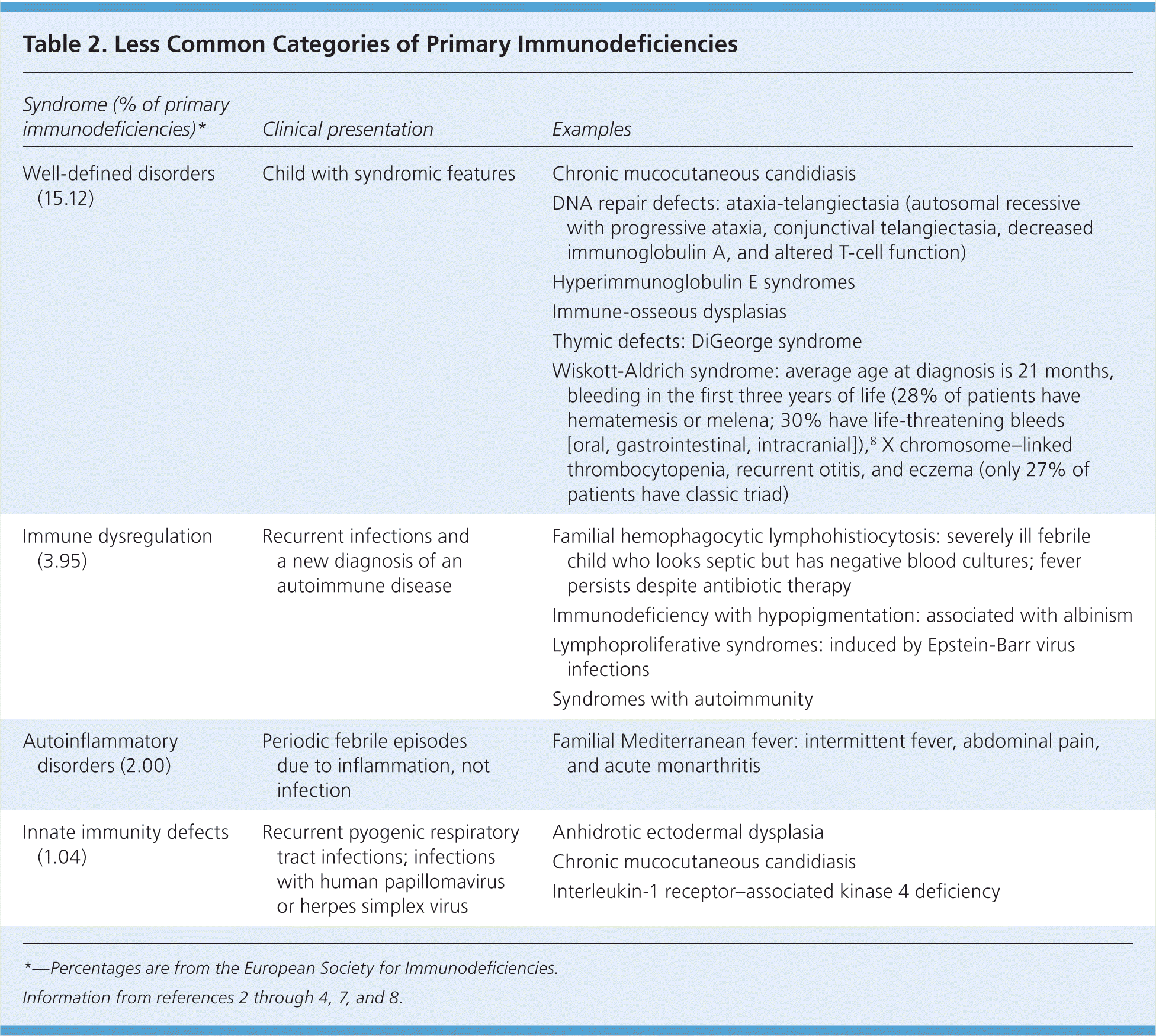
In the United States, the most common types of primary immunodeficiency disease in children are antibody disorders, followed by combined B-cell and T-cell disorders, phagocytic defects, and complement disorders.6 Less common categories of primary immunodeficiency diseases are reviewed in Table 2.2–4,7,8
Table 2. Less Common Categories of Primary Immunodeficiencies
| Syndrome (% of primary immunodeficiencies)* | Clinical presentation | Examples |
|---|---|---|
| Well-defined disorders (15.12) | Child with syndromic features | Chronic mucocutaneous candidiasis |
| DNA repair defects: ataxia-telangiectasia (autosomal recessive with progressive ataxia, conjunctival telangiectasia, decreased immunoglobulin A, and altered T-cell function) | ||
| Hyperimmunoglobulin E syndromes | ||
| Immune-osseous dysplasias | ||
| Thymic defects: DiGeorge syndrome | ||
| Wiskott-Aldrich syndrome: average age at diagnosis is 21 months, bleeding in the first three years of life (28% of patients have hematemesis or melena; 30% have life-threatening bleeds [oral, gastrointestinal, intracranial]),8 X chromosome–linked thrombocytopenia, recurrent otitis, and eczema (only 27% of patients have classic triad) | ||
| Immune dysregulation (3.95) | Recurrent infections and a new diagnosis of an autoimmune disease | Familial hemophagocytic lymphohistiocytosis: severely ill febrile child who looks septic but has negative blood cultures; fever persists despite antibiotic therapy |
| Immunodeficiency with hypopigmentation: associated with albinism | ||
| Lymphoproliferative syndromes: induced by Epstein-Barr virus infections | ||
| Syndromes with autoimmunity | ||
| Autoinflammatory disorders (2.00) | Periodic febrile episodes due to inflammation, not infection | Familial Mediterranean fever: intermittent fever, abdominal pain, and acute monarthritis |
| Innate immunity defects (1.04) | Recurrent pyogenic respiratory tract infections; infections with human papillomavirus or herpes simplex virus | Anhidrotic ectodermal dysplasia |
| Chronic mucocutaneous candidiasis | ||
| Interleukin-1 receptor–associated kinase 4 deficiency |
*—Percentages are from the European Society for Immunodeficiencies.
Antibody Disorders
Worldwide, antibody disorders are the most common type of primary immunodeficiency disease. Antibody disorders account for 55% of primary immunodeficiency diseases among patients who are entered in the ESID registry 4; in the United States, 78% involve an antibody disorder.6
Antibody disorders can be broadly characterized by the absence or presence of B cells. When B cells are present, disorders are further characterized by whether the B cells are of normal quantity or quality.9
Clinically, children who have an antibody disorder present with recurrent or severe bacterial infections of the ears, sinuses, and lungs, particularly with encapsulated organisms including Streptococcus pneumoniae and Haemophilus influenzae. Antibody disorders usually present three months after birth, once maternal immunoglobulin from placental transfer is gone.
AGAMMAGLOBULINEMIA
According to the ESID registry, agammaglobulinemia accounts for 13% of antibody disorders, with X chromosome–linked Bruton tyrosine kinase defect accounting for 84% of agammaglobulinemias.4 Infants who have agammaglobulinemia are born with a complete absence of B cells in their peripheral blood and in umbilical cord blood. On examination, the infant may have no tonsils or lymph nodes. On testing, all immunoglobulin subtypes are decreased, and there is an absence of B cells on lymphocyte subset analysis.
HYPOGAMMAGLOBULINEMIA
Hypogammaglobulinemia is characterized by low or deficient levels of any of the immunoglobulins (immunoglobulin A [IgA], IgE, IgG and IgG subclasses, IgM), or by an abnormal response of immunoglobulins to vaccinations. In the ESID registry, 82% of cases of antibody disorders involve a hypogammaglobulinemia, the most reported of which is common variable immunodeficiency, accounting for 46% of hypogammaglobulinemias.4 In common variable immunodeficiency, levels of at least two immunoglobulin types are low.
In the United States, IgA deficiency is the most common type of B-cell disorder at 30%, followed by IgG subclass deficiency (IgG2, IgG3, and IgG4) at 26%, hypogammaglobulinemia (including IgG1 subclass) at 23%, common variable immunodeficiency at 15%, and both transient hypogammaglobulinemia of infancy and a selective antibody deficiency at 3%.6 Reasons for the differences in the rates of antibody disorders between patients in the United States and Europe are unclear, but they primarily relate to differences in the ethnicities of populations, and differences in data collection.4
The nadir for IgG levels in infants occurs at three months of age, but a transient hypogammaglobulinemia can persist because of a prolonged nadir of IgG. Clinical infections during this time are usually mild. Serum IgG and IgA levels are decreased, but B-lymphocyte levels are normal on further testing.3,7
As with other antibody disorders, ear, sinus, and pulmonary infections are common; gastrointestinal problems such as diarrhea, malabsorption, and symptoms of irritable bowel syndrome also occur in children who have common variable immunodeficiency.8 Causative organisms for infection include Clostridium difficile, as well as species of Giardia, Salmonella, Campylobacter, and Yersinia.8
T-Cell Disorders
T-cell disorders account for 9% of primary immunodeficiency diseases in the ESID registry 4 and 10.5% in the United States.6 T-cell disorders are characterized by the absence or presence of T lymphocytes. Additionally, T cells are important to the normal functioning of B cells. As a result, most T-cell deficiencies lead to a combined T-cell and B-cell disorder.
T-cell disorders usually present early in life. The most serious form of T-cell disorder, severe combined immunodeficiency (SCID), presents in infants as an emergent condition with life-threatening infections. Diarrhea, failure to thrive, opportunistic infections, and severe routine infections in a child younger than three months should raise suspicion for SCID.
Phagocytic Disorders
Phagocytic disorders are the result of abnormalities in neutrophils or monocytes. These types of disorders account for 12.5% of primary immunodeficiency diseases in the ESID registry 4 and 8.5% in the United States.6
Chronic granulomatous disease is the most common phagocytic disorder in the ESID registry.4 It is usually diagnosed by five years of age, and is characterized by pneumonia, abscesses, suppurative adenitis, and gastrointestinal infections. The first manifestation may be omphalitis in infants.8
Other common presentations of chronic granulomatous disease (and of other phagocytic disorders) include recurrent pyogenic or fungal skin infections, or abscesses. Infections are related to the inability of the phagocytic system to kill catalase-positive organisms, including Staphylococcus aureus; Burkholderia cepacia; and Nocardia, Aspergillus, Serratia, and Candida species. Invasive fungal infection with disseminated Candida, Aspergillus, or Nocardia species,8,10 or invasive S. aureus or B. cepacia septicemia, should raise suspicion for a phagocytic disorder.7
Severe congenital neutropenia and leukocyte adhesion deficiency type 1 are phagocytic disorders that usually present within the first few weeks of life. Delayed separation of the umbilical cord (more than four weeks after birth), or erosive perianal ulcers, can be early signs of leukocyte adhesion deficiency type 1.3,7,8 Omphalitis can occur in severe congenital neutropenia and leukocyte adhesion deficiency type 1.
Complement Disorders
Complement disorders account for 2% of primary immunodeficiency diseases in the ESID registry 4 and 3% in the United States.6 More than 25 proteins are involved in the complement pathway, which complements the action of antibodies to destroy bacteria.11
These disorders involve infections with encapsulated organisms.7 A deficiency of C3 is associated with recurrent pyogenic infections with S. pneumoniae and H. influenzae.3,7 Deficiencies in C5 through C9 are associated with Neisseria meningitidis infections such as meningitis, sepsis, and arthritis.3,7
Clinical Presentation: Whom to Evaluate
The most common presentations of a primary immunodeficiency disease in children are recurrent ear, sinus, and pulmonary infections; diarrhea; and failure to thrive.3 These conditions are also common in children who do not have an immunodeficiency disease, which raises the question of how to identify children in need of evaluation.
A detailed evaluation from the United Kingdom found that the three most helpful warning signs for primary immunodeficiency disease were a positive family history (relative risk [RR] = 18; 95% confidence interval [CI], 8 to 45 for any type of primary immunodeficiency disease); a diagnosis of sepsis treated with intravenous antibiotics (RR = 5; 95% CI, 1.4 to 15 for phagocytic disorders); and failure to thrive (RR = 22; 95% CI, 8 to 60 for T-cell disorders).12 Some of the key findings that can help family physicians identify a child with a primary immunodeficiency disease are shown in Table 3.1,3
Table 3. Clinical Signs That Suggest a Primary Immunodeficiency Disease

Many primary immunodeficiency diseases are hereditary (most hereditary conditions are autosomal recessive inherited or X chromosome–linked).1 A child with recurrent serious infections who has a positive family history of these types of diseases or who is from an ethnicity associated with higher parental consanguinity (e.g., northern and sub-Saharan Africa; the Middle East; portions of western, central, and southern Asia13) should be screened for an immunodeficiency disease.
A child with recurrent infections in a single anatomic location is more likely to have an anatomic defect than an immunodeficiency. If recurrent infections are present in two or more sites, however, an immunodeficiency disease can be suspected. Furthermore, children tend to “outgrow” their infections, with fewer infections as they get older. An immunodeficiency should be considered if a child's infections increase in frequency or become more severe as he or she gets older.3
Recurrent, serious infections with common pathogens may be a sign of an immunodeficiency disease. Similarly, any unusual infections, including meningitis, sepsis, and fungal and opportunistic infections, should raise suspicion for an immunodeficiency disease.3
Initial Evaluation
A basic laboratory workup that includes testing for human immunodeficiency virus (HIV) antibody, complete blood count with differential, and measurement of serum immunoglobulin and complement levels can identify children who need further testing and referral to a subspecialist for a suspected immunodeficiency disease.3,7,10,12,14
HUMAN IMMUNODEFICIENCY VIRUS
HIV infection should be considered in newborns and adolescents who present with diarrhea, failure to thrive, and unusual opportunistic infections.3 HIV infection clinically resembles a T-cell immunodeficiency disorder. For children 18 months or older, HIV antibody testing for diagnosis is sufficient.
Because maternal antibodies to HIV cross the placenta, viral testing is required in children younger than 18 months. HIV DNA polymerase chain reaction testing or HIV RNA assay testing in newborns is recommended during the first 14 to 21 days of life, at one month of age, and again at four to six months of age to identify those who have perinatal-acquired HIV infection.15
COMPLETE BLOOD COUNT
A complete blood count with differential should be obtained to screen for a T-cell or phagocytic disorder. T-cell disorders are characterized by lymphocytopenia. Newborns usually have a lymphocytosis. An absolute lymphocyte count of less than 3,000 per mm3 in a newborn can be used as the cutoff for consideration of a T-cell disorder.2
Absolute lymphocyte count is age-dependent. In the appropriate clinical situation, if the absolute lymphocyte count is two standard deviations below the mean, a T-cell disorder may be considered.16 For example, an absolute lymphocyte count of less than 2,800 per mm3 in a four-month-old infant has 86% sensitivity and 94% specificity for detecting SCID.17
T-cell disorders can be further confirmed by lack of a delayed hypersensitivity skin test response to Candida, mumps, or tetanus in children older than one year, and by lymphocyte subset analysis at any age.14 A lymphocyte subset analysis will screen for the number and percentage of T cells (CD3, CD4, CD8), B cells (CD19, CD20), and natural killer cells (CD16, CD56).
Phagocytic disorders are characterized by neutropenia and abnormalities in lysosomal granules in neutrophils.14 Primary immunodeficiency disease should be suspected if the neutrophil count is less than 1,500 per mm3. If the neutrophil count is normal but there is still suspicion of primary immunodeficiency disease, granulocyte function tests can be performed.10
SERUM IMMUNOGLOBULINS
Patients with B-cell disorders have low serum immunoglobulin levels and decreased production or response of immunoglobulins to vaccination.7,14 Serum immunoglobulin levels vary with age, so age-specific cutoffs should be used when performing laboratory testing.18 If immunoglobulin levels are low, serum albumin level should be checked because low albumin suggests protein loss through the kidney or protein malabsorption in the bowel as causes of immunoglobulin deficiency.14
IgG antibody titers to vaccine antigens can be checked to determine responsiveness to vaccination. Testing is ideally performed four weeks after vaccination if the child was not previously exposed to the vaccine antigen. If the child was previously exposed, a threefold increase in the titers against two antigens that is present three weeks after vaccination indicates responsiveness of B cells.14 Protein antigens (tetanus, diphtheria, rubella, and mumps) can be checked at all ages; polysaccharide antigens (e.g., H. influenzae, Pneumococcus species) can be checked if the patient is two years or older.14 If the physician is using a conjugated pneumococcal vaccine, specific serotype IgG antibody titers are needed.7
COMPLEMENT TESTING
Complement disorders are screened by checking the components of the classic and alternative pathways. The classic pathway involves C1 through C9 and is checked with a CH50 assay. If the assay results are normal, the child does not have a clinically significant complement deficiency. If the results are abnormal, the alternative pathway should be checked with an AH50 or CH100 assay. Using the combination of the two test results, specific deficiencies can be identified.10,14
Newborn Screening for T-Cell Disorders
In 2010, the U.S. Department of Health and Human Services recommended routine screening for SCID in newborns.19 SCID is estimated to occur in one in 100,000 live births.3 Although SCID is rare, early identification and treatment with hematopoietic stem cell transplantation can prevent infant deaths. In the United States, the survival rate is 94% if transplantation is performed in the first three-and-a-half months of life, but drops to 70% if transplantation occurs later than that.20 Death is primarily related to viral illness present at diagnosis.20
Five states currently require screening for T-cell disorders in newborns; another 15 states are in the process of introducing testing.19 Screening involves detection of T-cell receptor excision circles by polymerase chain reaction using the current newborn heel stick dried blood spot.17 T-cell receptor excision circles are pieces of DNA produced only by T cells.
Using a cutoff of less than 30 copies per μL, screening for T-cell receptor excision circles is 100% sensitive for a T-cell disorder. The false-positive rate is 1.5% in term, well infants, and 5% in preterm infants or those who are in the intensive care unit.17 Testing protocols include resampling or first sampling at an estimated gestational age greater than 37 weeks.
No cases of SCID have been identified with current screening programs, but other T-cell disorders have been found. In Wisconsin, eight infants with T-cell disorders were noted out of 71,000 infants screened.21 In Massachusetts, 19 infants with T-cell disorders were identified out of 77,491 infants screened.22
Data Sources: An OVID Medline search was completed using the text word primary immunodeficiency with the MeSH term immunologic deficiency syndromes exploded. These results were then combined with various diagnostic terms and text words. The search included meta-analyses, randomized controlled trials, clinical trials, other studies, and reviews. Also searched were the Agency for Healthcare Research and Quality evidence reports, Clinical Evidence, DynaMed, EBM Reviews (all databases), the National Guideline Clearinghouse, PIER, and the U.S. Preventive Services Task Force. Search date: December 1, 2011.