Primary bone cancers include osteosarcoma, Ewing sarcoma, and chondrosarcoma. They account for less than 1% of diagnosed cancers each year and are associated with significant morbidity and mortality. Timely diagnosis is challenging because of late patient presentation, nonspecific symptoms that mimic common musculoskeletal injuries, and low suspicion by physicians. Plain radiography is the preferred diagnostic test. Radiographic suspicion of a bone malignancy should prompt quick referral to a cancer center for multidisciplinary care. Osteosarcoma, the most common bone cancer, most often occurs in children and adolescents. It typically develops in the metaphysis of long bones, specifically the distal femur, proximal tibia, and proximal humerus. Metastasis to the lungs is common. Use of neoadjuvant and adjuvant chemotherapy, in combination with surgery, has improved survival rates to nearly 80% for patients with localized disease, and 90% to 95% of patients do not require limb amputation. Ewing sarcoma is the second most common bone cancer and is similar to osteosarcoma in terms of presenting symptoms, age at occurrence, and treatment. Prognosis for osteosarcoma and Ewing sarcoma depends on the presence of metastasis, which lowers the five-year survival rate to 20% to 30%. Chondrosarcoma is the rarest bone cancer, primarily affecting adults older than 40 years. Survival rates are higher because most of these tumors are low-grade lesions.
Skeletal bones can host numerous types of cancers. They commonly receive metastases from other malignancies, including breast, lung, renal, prostate, and thyroid cancers. Bone marrow can be the nidus of malignancy in multiple myeloma, lymphoma, and leukemia. In all of these illnesses, however, the malignant cells are not of bony origin.
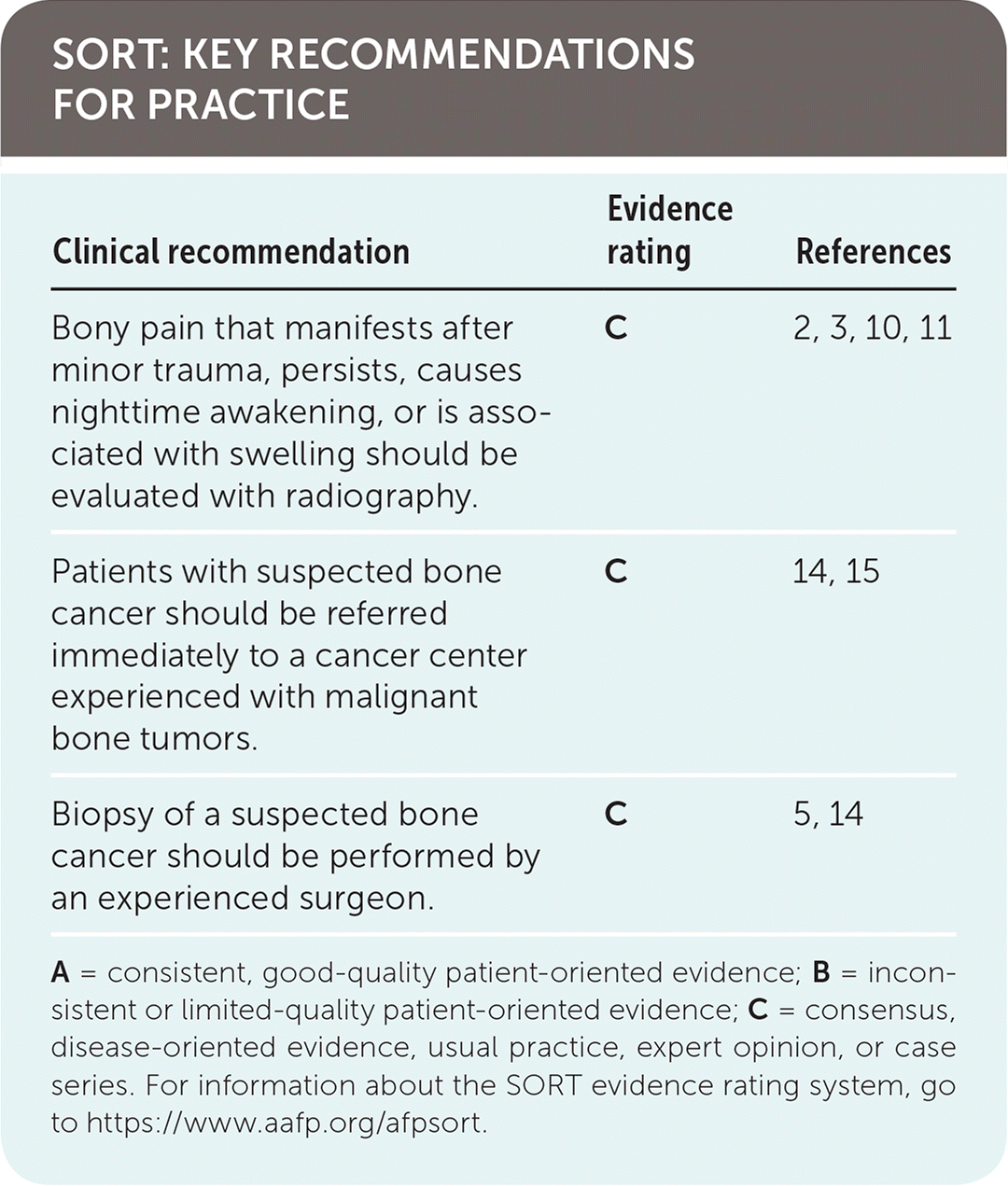
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Bony pain that manifests after minor trauma, persists, causes nighttime awakening, or is associated with swelling should be evaluated with radiography. | C | 2, 3, 10, 11 |
| Patients with suspected bone cancer should be referred immediately to a cancer center experienced with malignant bone tumors. | C | 14, 15 |
| Biopsy of a suspected bone cancer should be performed by an experienced surgeon. | C | 5, 14 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
Three types of cancers arise in bone itself: osteosarcoma, Ewing sarcoma, and chondrosarcoma. Although these malignancies account for less than 1% of all diagnosed cancers each year, their morbidity and mortality are significant.1 This article reviews the characteristics of primary bone cancers, their diagnosis, treatment, and prognoses.
Osteosarcoma
Osteosarcoma is the most common bone cancer, accounting for nearly two-thirds of all cases.2–4 Approximately 1,200 patients are diagnosed with osteosarcoma in the United States annually.1 Osteosarcoma is primarily an illness of childhood, with a small increase in incidence among persons older than 60 years. It is the third most common childhood malignancy, with a median incidence at 12 years of age for girls and 16 years for boys.2
Osteosarcoma is thought to originate from malignant primitive mesenchymal cells that differentiate into osteoblasts, which in turn produce a malignant osteoid matrix. Osteosarcomas can arise in any bone, but classically develop in the metaphyses of long bones. Nearly 60% occur in the distal femur, the proximal tibia, and the proximal humerus.3 A bone’s metaphysis contains the growth plate, which is responsible for active bone formation and elongation. Thus, osteosarcomas tend to occur at the age and location at which bony growth is the most active and when cells are vulnerable for mutations (Figure 1).
FIGURE 1.
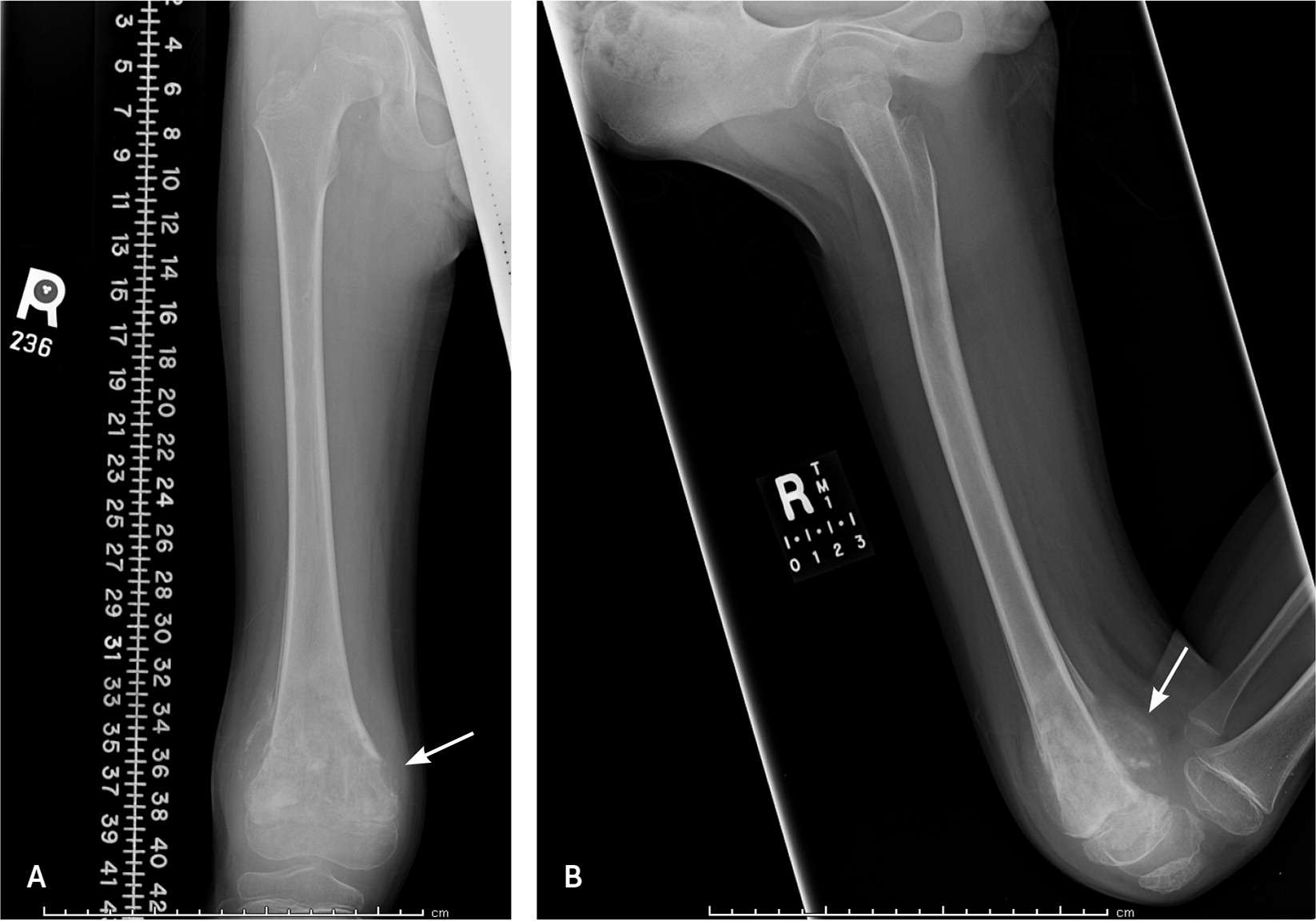
Radiographs of the distal femur of an eight-year-old boy with distal thigh pain and swelling that started after a fall at school two days previously. (A) Anteroposterior view shows a predominantly lucent, sclerotic lesion with significant periosteal reaction. (B) Lateral view demonstrates posterior soft tissue tumor involvement. Subsequent biopsy of the lesion confirmed high-grade osteosarcoma.
Osteosarcomas can metastasize regionally and systemically. Primary tumors can form “skip lesions” by intraosseous spread within the same bone or transarticular spread across a joint, such as between the tibia and femur.5 Systemic metastasis of osteosarcoma most often occurs in the lungs. Bones of another extremity are the second most common location of metastasis.
Ewing Sarcoma
Ewing sarcoma is the second most common type of bone cancer, comprising about one-third of cases in the United States.2–4 Its estimated incidence is one in 100,000 among persons 10 to 19 years of age. It is more common in whites and Asians than in blacks.4
The cell origin of Ewing sarcoma is not known. It has been hypothesized that these tumors derive from undifferentiated, primitive neuroectodermal or neural crest cells. Recently, it has been suggested that Ewing sarcoma originates from primitive stem cells, and the degree of malignancy depends on the stage of stem cell arrest during differentiation.6 Ewing sarcoma is included in a group of tumors known as small blue round cell tumors, based on their microscopic features.
Ewing sarcoma and osteosarcoma have similar characteristics. Ewing sarcoma primarily affects children and adolescents, with a median age of 15 years.2 Ewing tumors also classically metastasize to the lungs and other bones. A major difference is the anatomic locations in which Ewing sarcomas typically develop: the pelvis (Figure 2), diaphysis of long bones, ribs, and scapula.
FIGURE 2.
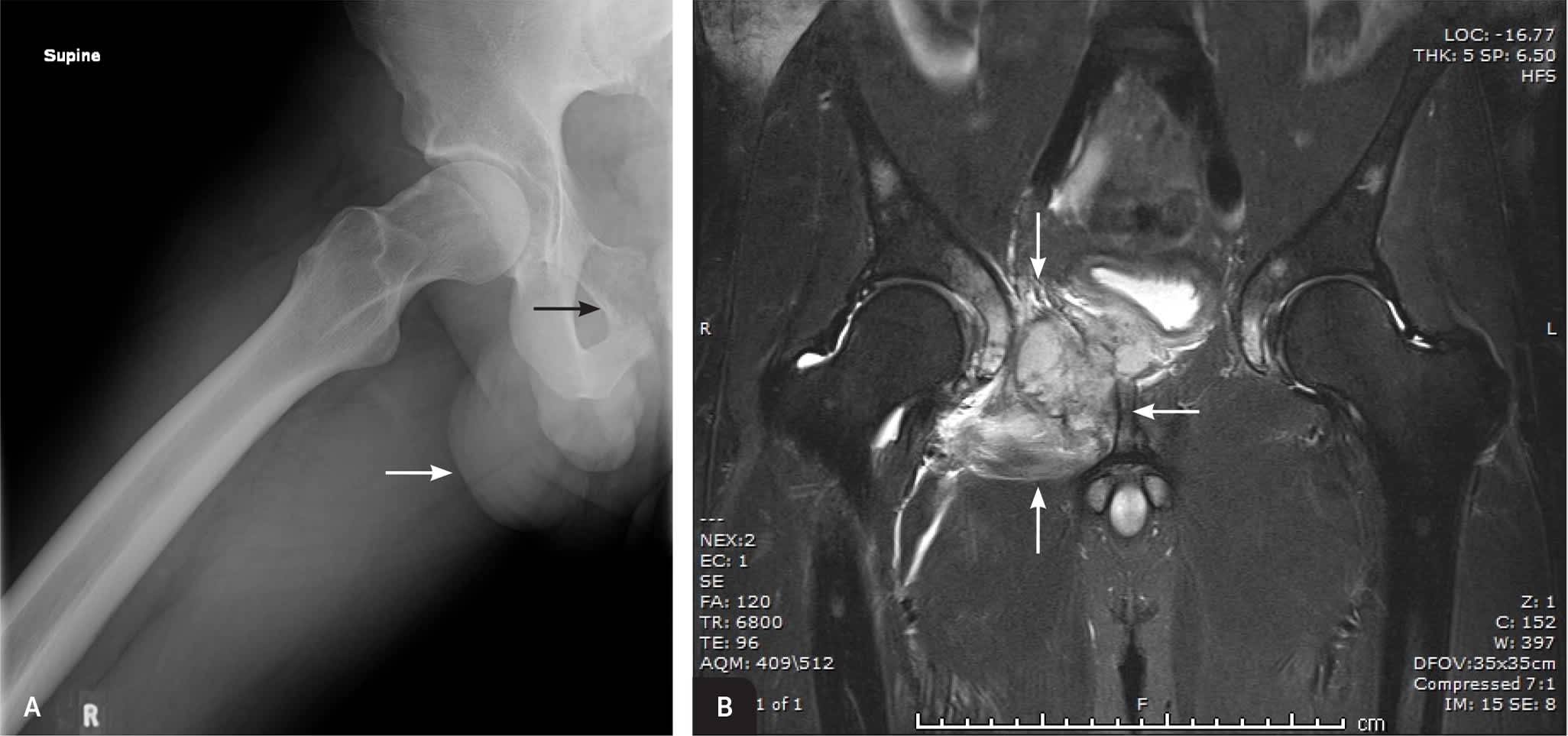
Imaging of the right hip of a 19-year-old man showing a destructive lesion of the right superior pubic ramus (black arrow) with an associated large anterior soft tissue mass (white arrow). (A) Radiography. (B) Magnetic resonance imaging immediately after radiography confirmed the bony superior pubic ramus tumor with soft tissue extension (arrows). Subsequent biopsy confirmed the diagnosis of Ewing sarcoma.
Chondrosarcoma
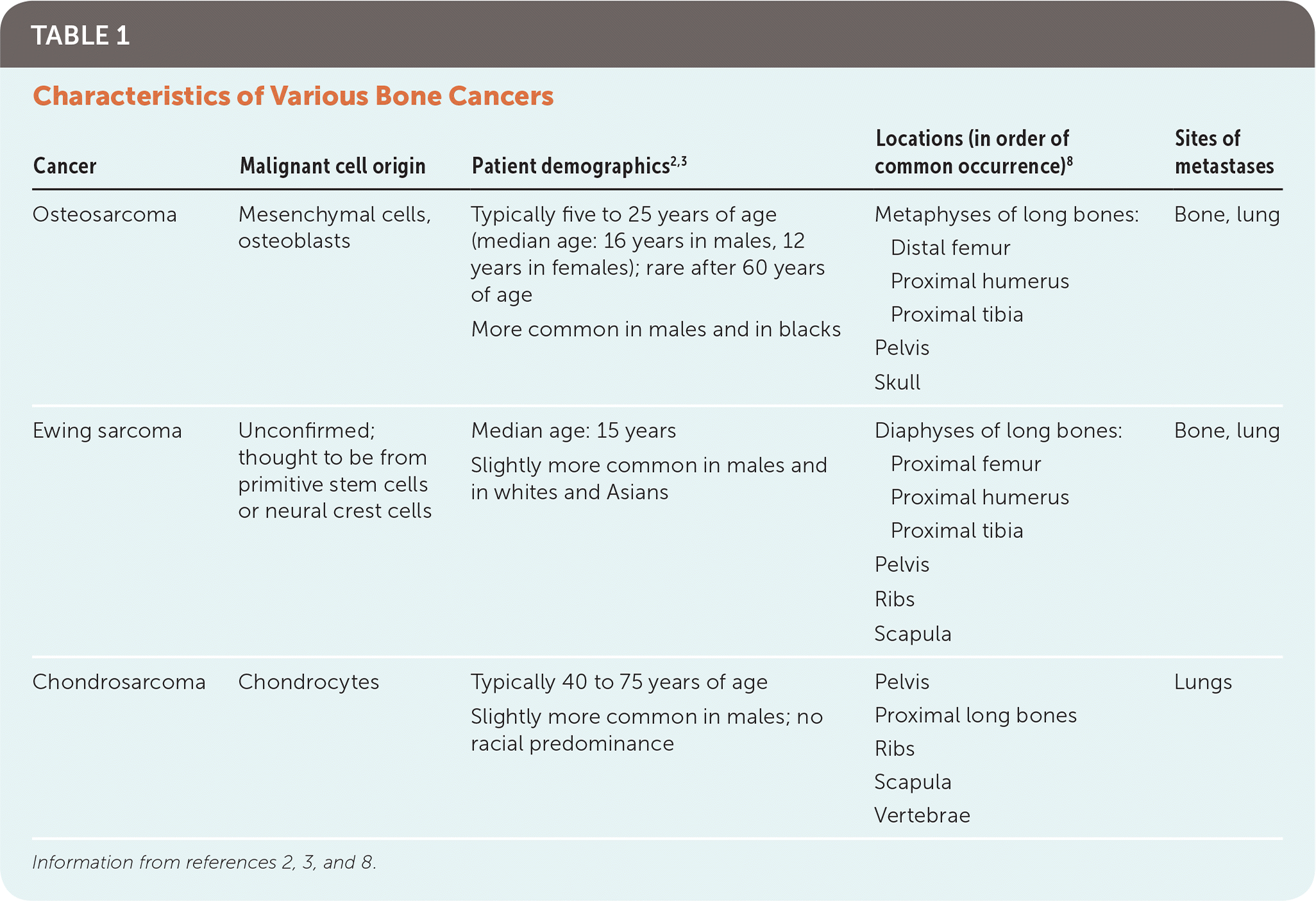
Chondrosarcoma is a malignant, cartilage-producing bone tumor. It is the least common bone cancer, with an estimated incidence of one in 200,000 persons.7 Unlike osteosarcoma and Ewing sarcoma, chondrosarcoma typically manifests in adults 40 to 75 years of age. It occurs more in the central skeleton, commonly arising from the pelvic girdle, vertebrae, and proximal long bones (Table 1).2,3,8
TABLE 1. Characteristics of Various Bone Cancers
| Cancer | Malignant cell origin | Patient demographics2,3 | Locations (in order of common occurrence)8 | Sites of metastases |
|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Diagnosis
The diagnosis of bone cancer can be challenging because of its low incidence and nonspecific symptoms at presentation. Timely diagnosis occurs when physicians consider bone cancer in the differential diagnosis and promptly obtain imaging.
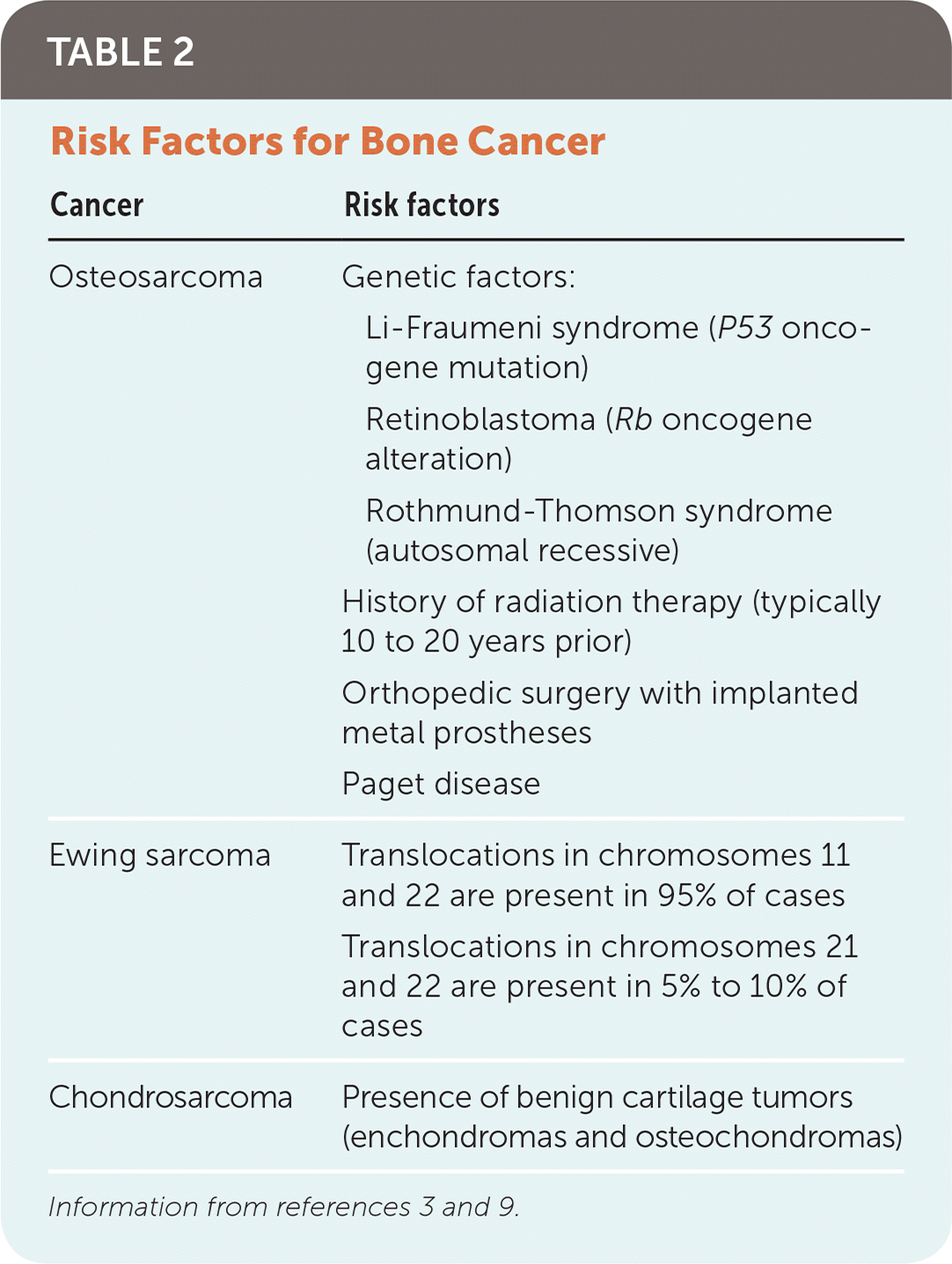
RISK FACTORS
Certain genetic syndromes increase the risk of osteosarcoma (Table 2).3,9 Patients with bilateral retinoblastoma, which arises from an alteration in the oncogene Rb, have a 40% chance of developing osteosarcoma by 40 years of age.3
TABLE 2. Risk Factors for Bone Cancer
| Cancer | Risk factors |
|---|---|
|
|
|
|
|
|
Clinical conditions that result in bone turnover increase the risk of bone cancer. Examples include metal implants within the bone from orthopedic interventions and Paget disease.9 Patients with a history of radiation therapy have a higher risk of osteosarcoma in previously irradiated bone. There are few known risk factors for Ewing sarcoma and chondrosarcoma, and they are not associated with familial cancer syndromes or radiation.
PRESENTING SYMPTOMS
Regional or localized pain with associated overlying tenderness and decreased range of motion are the most common presenting symptoms of bone cancer.10,11 These symptoms may mimic common musculoskeletal injuries, and pain often begins after minor physical trauma.
Soft tissue swelling and unexplained fevers are other common symptoms of bone cancer. Fever can relapse in patients with Ewing sarcoma, with intervals of weeks or months without symptoms. Nighttime pain and pathologic fracture can be concerning manifestations of bony malignancies; however, these symptoms are not common. Although bone cancer should be part of the differential diagnosis for patients with nighttime awakenings with bony pain, pain during the night is present in only 21% to 37% of patients with osteosarcoma and 19% of patients with Ewing sarcoma.11,12 Pathologic fracture is noted in 7% to 8% of patients with osteosarcoma.3,11
DIAGNOSTIC TESTING
Plain radiography is the preferred imaging modality for diagnosing malignant bone tumors.2,3,13 Patients commonly associate initial symptoms with minor trauma, and primary care physicians often order radiography of the affected limb with a clinical suspicion for fracture rather than bone malignancy. Table 3 lists common radiographic findings in patients with bone cancer.3,13 When radiographic findings raise suspicion for bone malignancy, laboratory studies (urinalysis, erythrocyte sedimentation rate, liver function testing, blood urea nitrogen, and creatinine level) should be obtained to evaluate other systems.
TABLE 3. Radiographic Characteristics of Bone Cancers

| Osteosarcoma |
| Dense sclerosis of metaphysis (nearly all) |
| Soft tissue extension (75%) |
| Radiating calcification; “sunburst” (60%) |
| Osteosclerotic lesion (45%) |
| Lytic lesion (30%) |
| Ewing sarcoma |
| Bone destruction (75%) |
| Soft tissue extension (64%) |
| Reactive bone formation (25%) |
| Lamellated periosteal reaction; “onion skin” (23%) |
| Radiating calcification; “sunburst” (20%) |
| Radiography may be normal |
| Chondrosarcoma |
| Calcified eccentric osteolytic lesion |
Radiographs showing potential malignancies should prompt additional imaging (Figure 3 and Figure 4). Early magnetic resonance imaging is superior to computed tomography for staging of bone tumors before treatment and can help determine tumor involvement in surrounding muscle, soft tissue, and neurovascular structures—information that is critical for limb-salvage surgery.13 Bone scans and chest computed tomography are useful in evaluating for metastasis.3 The role of positron emission tomography in the initial workup of bone cancer has yet to be established, but it is useful in evaluating for metastases and tumor recurrence. When combined with computed tomography, positron emission tomography can be used to plan biopsies or evaluate lesions in patients who cannot undergo magnetic resonance imaging.13
FIGURE 3.
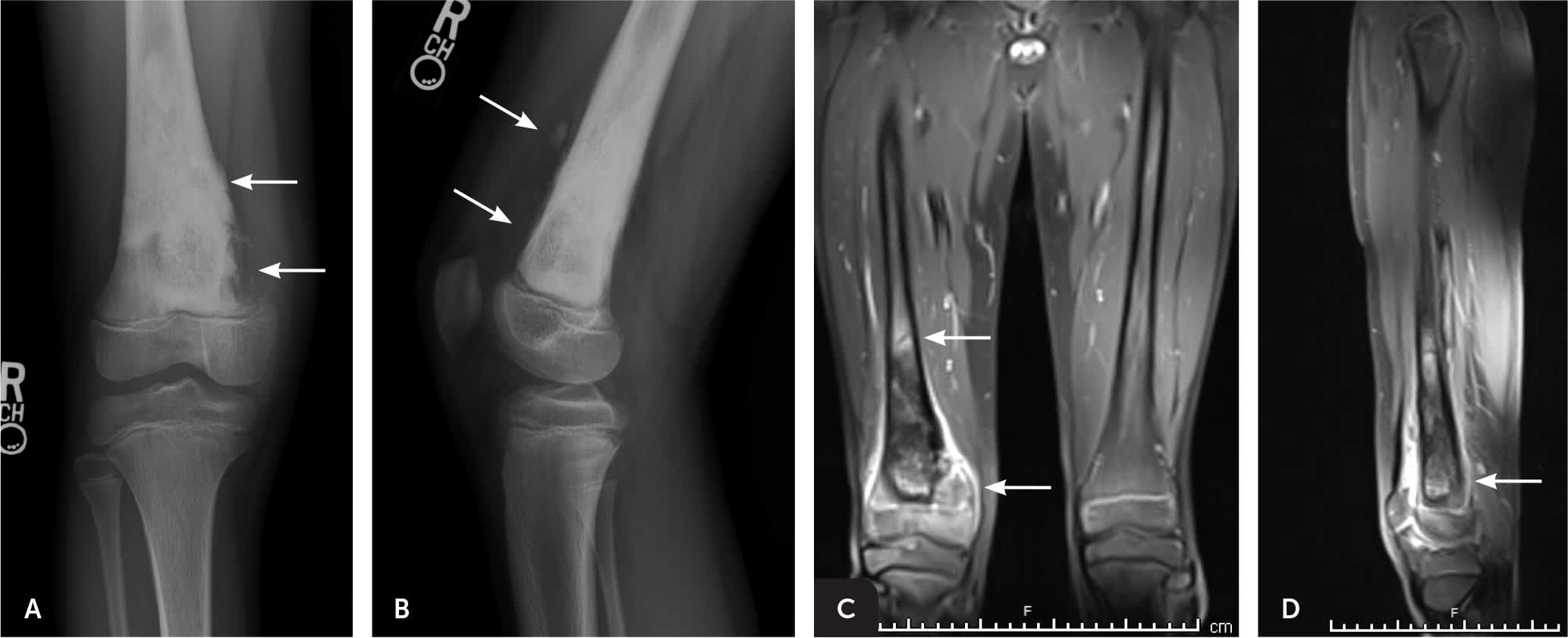
Imaging of the right knee of an 11-year-old boy with right knee pain for approximately four weeks. Although there was no history of specific trauma, the patient and his parents attributed the pain to daily participation in basketball. (A) The anteroposterior view shows an ill-defined mixed sclerotic and lytic lesion in the distal right femoral metaphysis. (B) The lateral view shows anterior soft tissue swelling and osteoid matrix deposition. (C) Coronal magnetic resonance imaging after radiography better defines the extent of tumor involvement. (D) Sagittal magnetic resonance imaging better shows the anterior soft tissue tumor extension. Subsequent biopsy of the lesion confirmed high-grade osteosarcoma.
FIGURE 4.
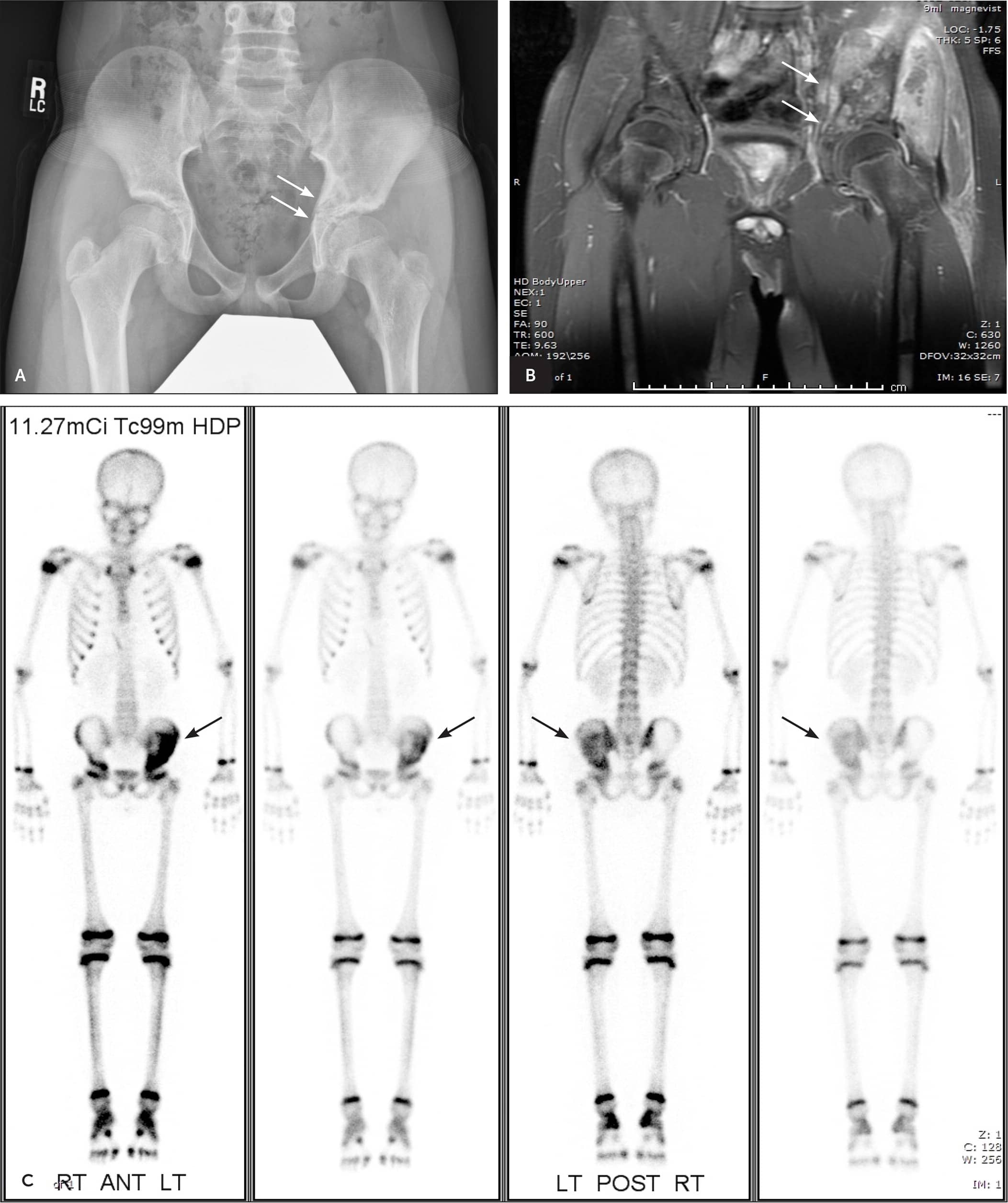
Imaging of the pelvis of an 11-year-old boy with left hip pain one week after falling during a football game. (A) The anteroposterior view shows asymmetric patches of lucency and sclerosis in the inferomedial left iliac bone. (B) Coronal magnetic resonance imaging shows a large, hyperintense, T2, aggressive-appearing enhancing mass with an extensive soft tissue component. The mass measured approximately 18.7 cm × 8.7 cm × 10.2 cm. (C) A whole-body nuclear bone scan shows significant signal intensity of the left iliac bone but does not suggest bony metastases at the initial presentation. Subsequent biopsy confirmed Ewing sarcoma of the iliac bone.
AVOIDING DIAGNOSTIC DELAYS
Because of the aggressive nature of bone cancers, early diagnosis is critical. However, delays in diagnosis are common.11 In the absence of trauma or known injury, a patient’s perception of symptoms may not elicit initial concern. Bone cancers can also present with relapsing and remitting symptoms, including pain and fever. Symptom-free periods may mislead physicians and patients into thinking the condition has resolved. Initial pain and swelling in bone cancers can mimic symptoms of minor musculoskeletal injuries.
In cases of persistent or recurrent bony pain or soft tissue swelling without known trauma, physicians should be encouraged to perform radiography. In one study, the presence of a palpable mass and the physician’s decision to order radiography were associated with a shorter diagnostic delay.11
Common Principles of Treatment
REFERRAL TO CANCER CENTER
Although treatment of bone cancers varies in the type of modalities used, the main factor in maximizing survival and quality of life is expedient referral to a cancer center at the time a bone malignancy is suspected.14,15 Because bone cancers most often occur in children and adolescents, children’s cancer hospitals typically have more experienced treatment teams. A multidisciplinary team includes a primary care physician, an orthopedic surgical oncologist, a pathologist experienced in bone tumors, pediatric oncologists, rehabilitation specialists, pediatric nurse specialists, and social workers.
BIOPSY
A properly performed biopsy starts the process of confirming the diagnosis of bone cancer, establishes tumor grade, and directs treatment. An experienced surgeon should perform the procedure.14 Whether the biopsy is obtained via open surgical technique or by percutaneous needle, biopsy tracts and skin can become contaminated with malignant cells. Thus, biopsy sites require complete subsequent removal (en bloc) and should be planned carefully to maximize the opportunity for limb salvage. Improperly performed biopsies can lead to misdiagnosis, local recurrence, amputation, and decreased survival.5
Treatment of Osteosarcoma
Before 1970, surgical excision was the only treatment for osteosarcoma. Amputations of extremities were common to achieve clear margins, and the five-year survival rate for patients with classic high-grade tumors was less than 20%.16–18 Over the next two decades, neoadjuvant (preoperative) and adjuvant (postoperative) chemotherapy were proven effective.17 The current mainstay of treatment for osteosarcoma is neoadjuvant chemotherapy, followed by surgical resection and adjuvant chemotherapy. Radiation therapy is less effective and is seldom used. The current five-year survival rate is more than 70% in persons with nonmetastatic disease.17
CHEMOTHERAPY
Neoadjuvant chemotherapy regimens aim to cause tumor necrosis and decrease primary tumor size, as well as the number and size of pulmonary metastases.17–19 It has increased the feasibility of limb-salvage surgery by reducing the amount of tissue needed to achieve wide margins. Adjuvant chemotherapy has been successful in decreasing postsurgical metastasis. Chemotherapy drugs with proven effectiveness against osteosarcoma include high-dose methotrexate, doxorubicin (Adriamycin), and cisplatin.3
SURGERY
Surgical excision is the definitive treatment for osteosarcoma. The goal of resection is to remove primary tumors with clear margins to limit recurrence and metastasis. Currently, 90% to 95% of patients with extremity osteosarcoma avoid amputation with successful limb-sparing resections.5 The success of limb-salvage surgery is largely due to improved preoperative planning with magnetic resonance imaging, advances in soft tissue and vascular surgery techniques, and enhanced fabrication and expansion of endoprostheses.19 Limb amputations are still necessary for some large tumors, cases in which vital vascular structures are compromised, and for poor response or tumor progression during chemotherapy.20
METASTASES
Osteosarcoma most often metastasizes to the pulmonary system through blood; 20% of patients initially present with evidence of lung metastases on computed tomography.16 As with primary lesions, treatment of metastases relies on chemotherapy. However, induction chemotherapy often does not resolve metastases entirely. Surgical resection of metastatic lung lesions is recommended, unless the location or amount of involved tissue makes surgery clinically unfeasible.16,20
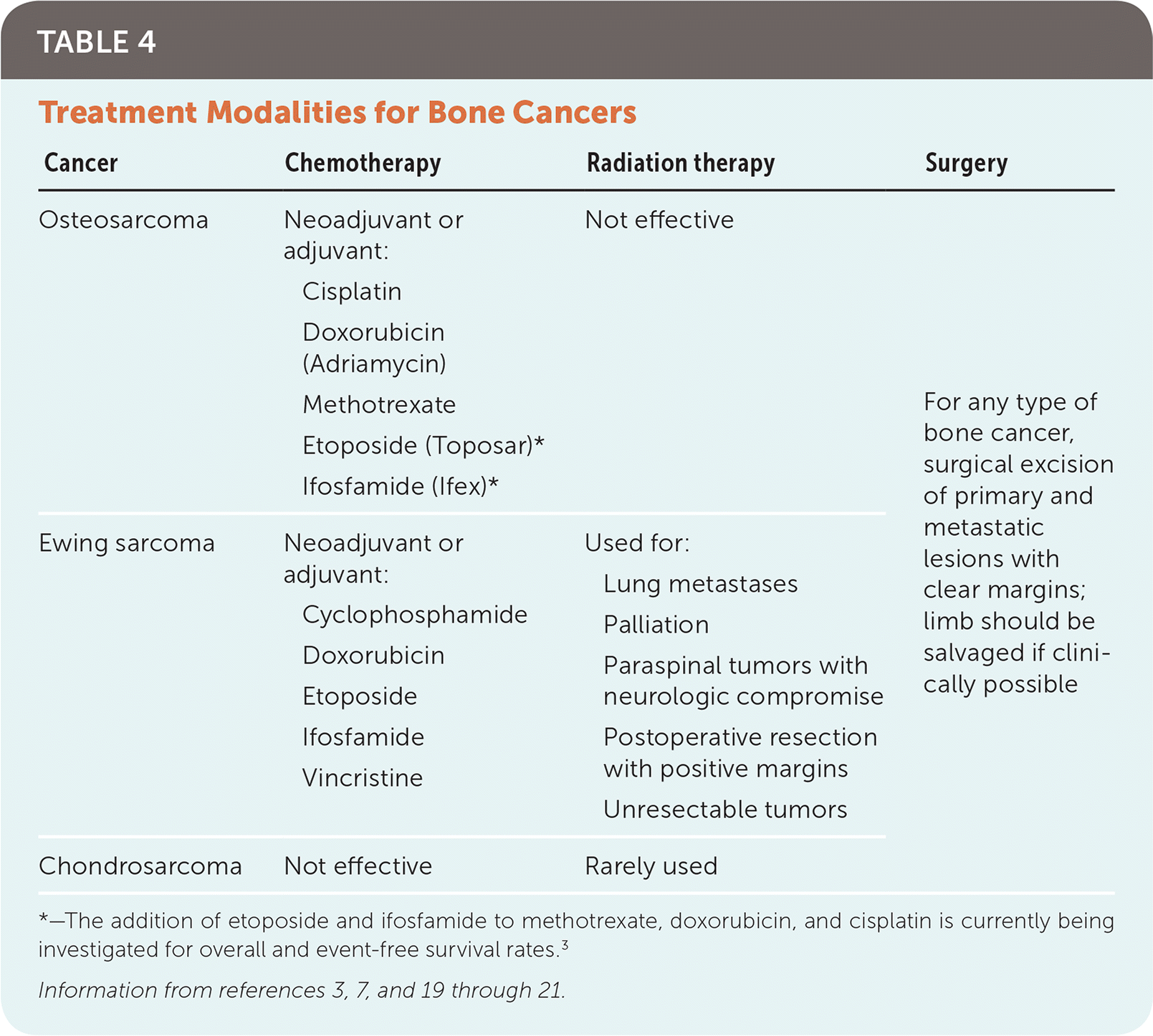
Treatment of Ewing Sarcoma
Disease-specific chemotherapy before and after surgical excision is standard for local and metastatic disease (Table 4).3,7,19–21 A major difference in the treatment of Ewing sarcoma compared with osteosarcoma is the use of radiation therapy as an alternative to surgery. Radiation therapy is used for primary Ewing lesions that are not amenable to surgical resection because of tumor location or size, when surgical margins are positive for residual tumor, when neurologic compromise is present from spinal tumors, and for lung metastases. When possible, surgery is typically preferred because clear-margin excisions have improved survival rates in Ewing sarcoma. Radiation is also thought to inflict greater long-term morbidity by retarding childhood bone growth, and radiation is associated with the development of subsequent neoplasms.4,22
TABLE 4. Treatment Modalities for Bone Cancers
| Cancer | Chemotherapy | Radiation therapy | Surgery |
|---|---|---|---|
|
|
|
|
|
|
| |
|
|
|
*—The addition of etoposide and ifosfamide to methotrexate, doxorubicin, and cisplatin is currently being investigated for overall and event-free survival rates.3
Treatment of Chondrosarcoma
Unlike osteosarcoma and Ewing sarcoma, chondrosarcoma is generally resistant to chemotherapy. It is thought that malignant cartilage cells have limited vascular connections, making delivery of chemotherapeutic agents ineffective.2,7 Surgical resection is the primary treatment for primary and metastatic chondrosarcoma. Similar to the treatment of Ewing sarcoma, radiation therapy is reserved for cases when adequate surgical margins cannot be achieved.
Prognosis
Prognosis varies greatly among the three types of bone cancers and is influenced by many variables (Table 5).2,3,7,21,23 The most significant prognostic indicator for osteosarcoma and Ewing sarcoma is the presence of metastases at the time of diagnosis. Patients with metastatic osteosarcoma or Ewing sarcoma have a survival rate of only 20% to 30%, compared with 70% to 80% for localized disease.16,21,23 Response to neoadjuvant chemotherapy is another significant prognostic marker for osteosarcoma and Ewing sarcoma. Patients with osteosarcoma who have necrosis of more than 90% of the primary tumor on histologic review of surgical tissue have an increased 10-year survival rate compared with patients with less response.17,21 The major prognostic factor for patients with chondrosarcoma is the grade of the tumor. Approximately 85% of these tumors are low grade, and overall survival is favorable.3
TABLE 5. Prognostic Factors for Bone Cancers
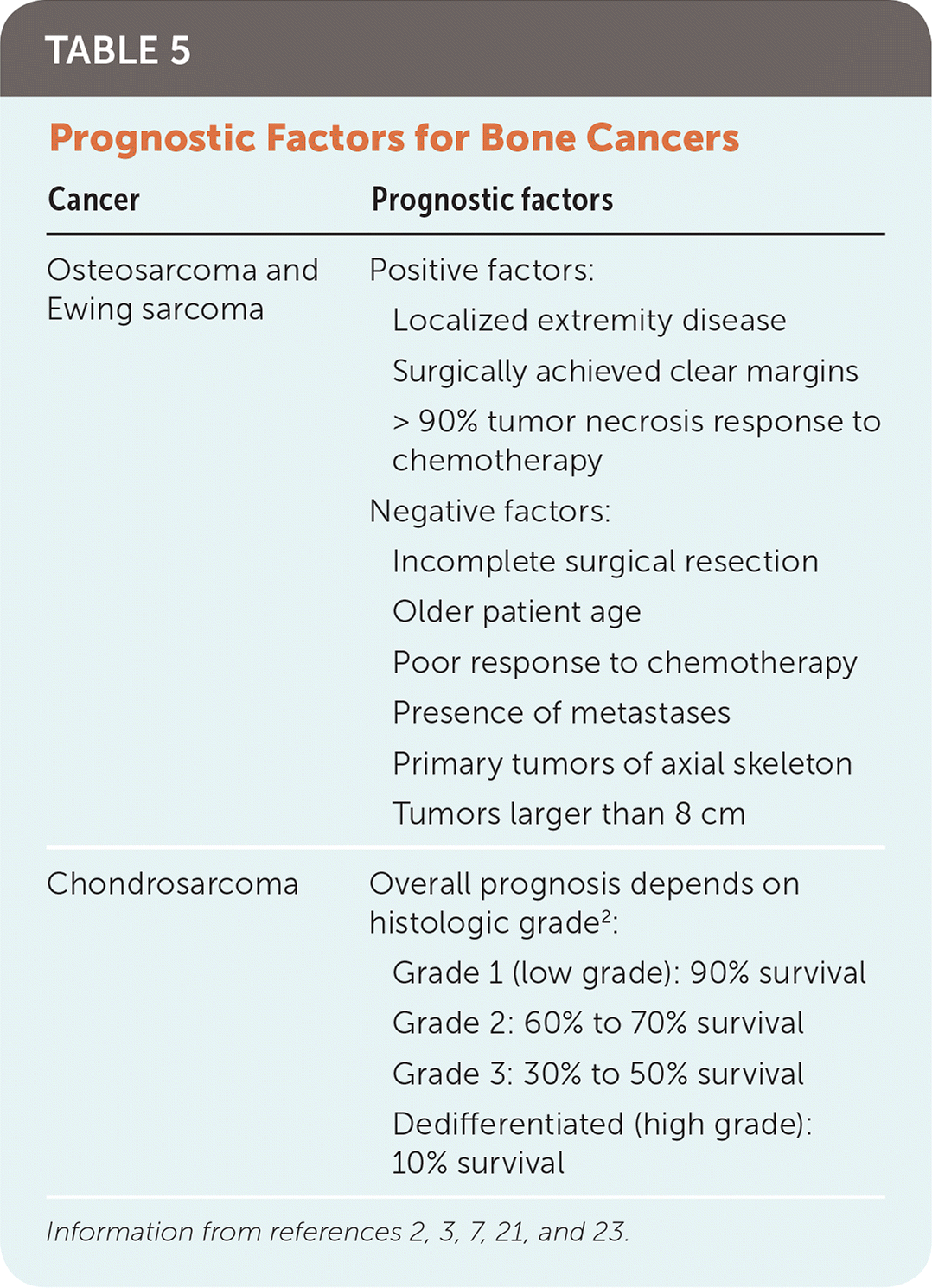
| Cancer | Prognostic factors |
|---|---|
|
|
|
|
Data Sources: A PubMed search was completed using the key terms bone cancer; bone metastasis; bone neoplasms; neoplasms, bone tissue; osteosarcoma; Ewing sarcoma, and chondrosarcoma. The search included meta-analyses, randomized controlled trials, clinical trials, and reviews. We also searched the Agency for Healthcare Research and Quality evidence reports, the Cochrane database, Essential Evidence Plus, the National Institute for Health and Care Excellence, and the National Cancer Institute. Search dates: December 29, 2016, to June 27, 2018.
The views expressed in this article are those of the authors and do not reflect the policy or position of the U.S. Army Medical Department, Department of the Army, Department of Defense, or the U.S. government.
