Cutaneous vascular lesions are the most common pediatric birthmarks. Flat vascular malformations tend to persist, but raised vascular lesions, known as hemangiomas, generally involute. Although not always necessary, treatment of flat lesions, if desired, is best accomplished with flash-lamp pumped pulsed dye laser. Therapy of hemangiomas varies depending on the presence of associated symptoms or syndromes. Specifically, hemangiomas that are likely to lead to loss of function or life (e.g., lesions of internal organs, lesions associated with coagulopathy) should be treated promptly. Treatment may also be required for hemangiomas that are likely to lead to scarring when the lesion involutes, such as hemangiomas of the nose and lip. The natural history of hemangiomas includes proliferative, stationary and involutional phases. Many superficial hemangiomas resolve with minimal sequelae.
Vascular birthmarks are commonly encountered in children and are classified as either hemangiomas or vascular malformations1,2 (Table 1). Hemangiomas are benign neoplastic proliferations of vascular endothelial cells characterized by spontaneous involution. In contrast, vascular malformations are not neoplasms but permanent morphogenic abnormalities of capillaries, veins, arteries or lymphatic vessels. Flat lesions (vascular malformations) tend to persist, while raised lesions (hemangiomas) tend to regress.
TABLE 1 Classification of Vascular Lesions
| Vascular malformations (flat lesions) |
| Salmon patch (also known as nevus simplex or nevus telangiectaticus) |
| Port-wine stain (also known as nevus flammeus) |
| Hemangiomas (raised lesions) |
| Superficial hemangioma (also known as capillary nevus hemangioma) |
| Deep hemangioma (also known as cavernous hemangioma) |
Vascular Malformations
Although vascular malformations are by definition present at birth, they may become clinically apparent at different ages. Capillary and lymphatic malformations are generally noted at birth; arterial and venous malformations become visible any time from birth to adulthood. Unlike hemangiomas, capillary and venous malformations are soft, flat, easily compressible and easily emptied of blood by manual pressure.
Salmon Patch (Nevus Simplex, Nevus Telangiectaticus)
The salmon patch (often called a “stork bite”), composed of dilated dermal capillaries, is the most common vascular malformation of infancy. It is present as a light-red to pink patch in approximately 70 percent of white neonates and approximately 60 percent of black neonates.2 Forty percent of all newborns exhibit these lesions on the nape of the neck; 20 percent have lesions on the upper eyelid or glabella. Although most eyelid lesions fade by six to 12 months of age and glabellar lesions by five to six years of age, 50 percent of nuchal-region salmon patches, also called Unna's nevus, persist into adulthood3 (Figures 1a and 1b).
FIGURE 1A.
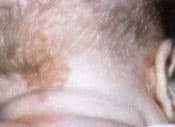
Salmon patch on the nape of the neck in a child. This lesion (termed “stork bite”) occurs in 40 percent of all newborns. Fifty percent of the lesions persist into adulthood (Figure 1b) but are usually covered by hair.
FIGURE 1B.
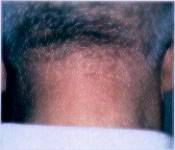
Salmon patch on the nape of the neck in an adult.
Treatment
Watchful waiting is usually recommended. Parents must be informed that in fair-skinned children, erythema may persist or reappear during episodes of crying, physical exertion or breath-holding. If therapy is desired, the flash-lamp pumped pulsed dye laser (FPDL) is the treatment of choice for these superficial vascular lesions.
Port-Wine Stain (Nevus Flammeus)
Occurring in 0.5 percent of newborns, port-wine stains are present at birth, persist throughout life and appear as pale pink to red-purple, usually unilateral macules of the face or extremities (Figure 2). The capillary ectasias that make up port-wine stains involve not only the superficial capillaries that are involved in the salmon patch, but also the deeper vessels of the dermis and subcutaneous tissue. Soft tissue or bony hypertrophy may occur either in an isolated port-wine stain or in Sturge-Weber syndrome or Klippel-Trénaunay-Weber syndrome.
FIGURE 2.
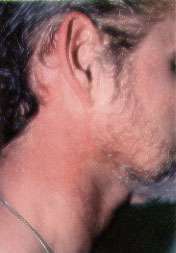
Port-wine stain persisting into adulthood.
Sturge-Weber Syndrome. Sturge-Weber syndrome is characterized by a port-wine stain in the distribution of the ophthalmic branch of the trigeminal nerve, with associated seizures (80 percent), mental retardation (60 percent), hemiplegia (30 percent) and neonatal glaucoma.3 Port-wine stains involving the region above the palpebral fissure are characteristic of Sturge-Weber syndrome (Figures 3a and 3b). From 5 to 8 percent of patients with a port-wine stain in the ophthalmic distribution of the trigeminal nerve develop Sturge-Weber syndrome; bilateral involvement or lesions of both the upper and lower eyelids increase the likelihood of this condition.
FIGURE 3A.
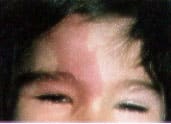
Port-wine stain in the ophthalmic distribution of the trigeminal nerve in a pediatric patient with Sturge-Weber syndrome, illustrating characteristic involvement of the upper eyelid.
FIGURE 3B.
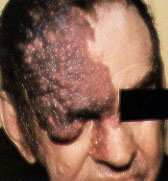
Note the soft tissue hypertrophy and overlying textural change in the port-wine stain in an adult shown here compared with a port-wine stain in a child shown in Figure 3a.
The associated vascular malformation of the ipsilateral leptomeninges and cerebral cortex produces seizures during the first year of life. Skull radiographs showing double-contoured parallel streaks of calcification may confirm the diagnosis, but computed tomographic (CT) scans or magnetic resonance images are more sensitive in detecting early calcifications. Patients suspected of having Sturge-Weber syndrome should undergo cerebral imaging studies, ophthalmologic examination and regular follow-up.
Klippel-Trénaunay-Weber Syndrome. Klippel-Trénaunay-Weber syndrome consists of a port-wine stain over an extremity, with associated soft tissue and bony hypertrophy that may result in hypertrophy of the extremity (Figure 4). An arteriovenous fistula is present in 25 percent of these patients; the condition is then referred to as Parkes-Weber syndrome. In cases of suspected Klippel-Trénaunay-Weber syndrome, the length and girth of the extremity should be measured every three to six months and, if elongation of an extremity is noted, imaging studies to evaluate the patient for the presence of an arteriovenous fistula or venous atresia should be performed. Surgical vascular intervention remains controversial.4
FIGURE 4.
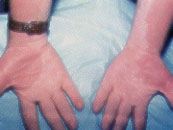
Port-wine stain over both arms in a patient with the associated soft tissue and bony hypertrophy of Klippel-Trénaunay-Weber syndrome.
Treatment
As patients with port-wine stains mature, the lesions become a deeper purple, and angiomatous bleeding papules and soft tissue hypertrophy often develop (Figure 5). Because of the propensity of these cutaneous lesions to persist and grow, treatment of cosmetically or functionally impairing port-wine stains is recommended during infancy to prevent the soft tissue and bony changes.5 Ninety-four percent of port-wine stains can be treated successfully with FPDL, which emits short pulses of concentrated energy in the wavelength of oxyhemoglobin and induces selective thermolysis of the cutaneous vasculature.6 This method results in minimal scarring and is the treatment of choice for flat vascular lesions. As an adjunct to therapy, opaque make-up may be used either indefinitely or while awaiting definitive treatment.
FIGURE 5.
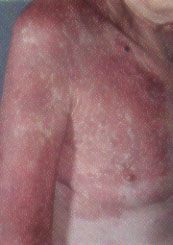
Port-wine stain in an adult, containing numerous angiomatous papules.
Hemangiomas
Epidemiology of Hemangiomas
Hemangioma is the most common benign neoplasm of infancy, occurring in 1 to 3 percent of newborns. Two types are recognized: the superficial strawberry or capillary type, which includes about 65 percent of all hemangiomas, and the deeper, cavernous type, which accounts for about 15 percent of hemangiomas. The remaining hemangiomas are mixed, with both superficial and deep components. The incidence of hemangiomas is elevated in female infants (two to five times higher than in male infants) and premature infants (especially those weighing less than 1,500 g [3 lb, 4.5 oz]).7
Hemangiomas may appear as telangiectatic macules or blanched spots that may not be recognized at birth. Skin ulceration may, rarely, be the presenting sign of hemangioma in the neonatal period.8 Ninety percent of hemangiomas are detected in the first month of life and may occur on the head and neck (60 percent), trunk (25 percent) and extremities (15 percent). Lesions that penetrate the superficial dermis appear as bright-red, well-defined nodules and are known as superficial hemangiomas (Figures 6 and 7). Deep hemangiomas of the dermis or subcutaneous tissues have less distinct borders and appear as slightly raised, bluish nodules (Figures 8 and 9).
FIGURE 6.
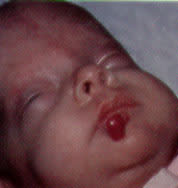
Superficial hemangioma in an infant, with the usual strawberry-red color.
FIGURE 7.
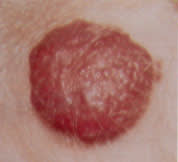
Well-defined, lobulated tumor in an infant, with minute surface capillaries characteristic of superficial hemangioma.
FIGURE 8.
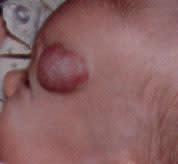
Bluish-red nodule typical of a mixed hemangioma.
FIGURE 9.
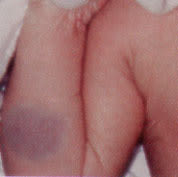
Deep hemangioma, seen as a poorly-defined, slightly raised, blue subcutaneous nodule with normal overlying skin.
Natural History of Hemangiomas
Nodules develop by two to four weeks of age, marking the six- to 12-month proliferative phase characteristic of hemangiomas. By one year of age, most hemangiomas have achieved their maximum size, ranging from 2 to 20 cm (average: 2 to 5 cm). A stationary phase predominates until 15 months of age. The involuting phase then begins, with the development of pale gray regions within the nodules and diminished firmness (Figures 10 and 11). Full regression occurs in 50 percent of patients by age five, in 70 percent by age seven and in 90 percent by age nine. Although superficial hemangiomas usually resolve with minimal atrophy, deep or mixed-type hemangiomas often show incomplete involution, with residual atrophic, wrinkled, telangiectatic, redundant skin (Figure 12). Normal skin is restored in only 50 percent of all hemangiomas.9
FIGURE 10.
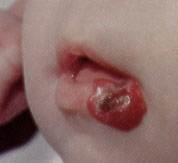
Involuting hemangioma, characterized by the development of pale gray regions, ulceration and bleeding.
FIGURE 11.
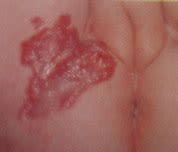
Involuting hemangioma, with pale gray regions, diminished firmness and minimal residual scarring.
FIGURE 12.
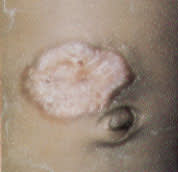
Spontaneous involution of a hemangioma, with prominent residual scarring.
Special Syndromes
Hemangiomas may exist in the setting of special syndromes, and the astute clinician may diagnose these variants early in their presentation.
Kasabach-Merritt Syndrome
A patient developing sudden enlargement and firmness in a preexisting large hemangioma, with easy bruising and petechiae, should be suspected of having Kasabach-Merritt syndrome, a coagulopathy characterized by thrombocytopenia occurring in patients with large hemangiomas. Platelet trapping within the enlarging hemangioma may result in secondary disseminated intravascular coagulation. These patients require hospitalization and treatment with prednisone (2 to 4 mg per kg per day) or interferon alfa-2a (Roferon-A) in steroid-resistant patients. External compression bandages and transfusion of supportive blood products facilitate recovery. If hemorrhage is prevented, recovery is usually complete.3
Diffuse Neonatal Hemangiomas
Widely disseminated, 2 to 15 mm, red to blue-black papular hemangiomas present at birth should alert the clinician to the possibility of associated visceral hemangiomas in the syndrome of diffuse neonatal hemangiomatosis. Gastrointestinal, hepatic, central nervous system and pulmonary visceral hemangiomas may be present, and high-output cardiac failure may occur by two to nine weeks of age. Evaluation of these patients includes a complete blood count, auscultation for bruits, liver edge palpation, ultrasonographic or CT scans of suspicious areas and evaluation of urine and stool for occult blood. Treatment includes hepatic lobectomy (if the liver is involved), vessel ligation or embolization, high-dose corticosteroids (2 to 4 mg per kg per day), interferon alfa-2a and treatment for high-output cardiac failure. Lesions spontaneously involute in some patients.
Benign Neonatal Hemangiomatosis
Cases of widespread cutaneous hemangiomas without visceral involvement are classified as benign neonatal hemangiomatosis. Extensive facial hemangiomas may be associated with intracranial involvement, and evaluation by CT or magnetic resonance imaging is mandatory to determine the presence of structural abnormalities of the brain.
Blue Rubber Bleb Nevus Syndrome
Infants with multiple cutaneous cavernous hemangiomas may have concurrent gastrointestinal tract hemangiomas in the blue rubber bleb nevus syndrome. Painful, soft, compressible, 1.0 to 50 mm blue rubbery nodules with a wrinkled surface are present at birth and increase both in size and number with age.10 Manual compression of these lesions expresses blood, leaving behind an empty wrinkled sac that rapidly refills. Small intestinal or colonic hemangiomas may be suspected in patients with anemia or positive stool guaiac specimens. Surgical excision of symptomatic cutaneous or gastrointestinal lesions remains the treatment of choice. Sclerosing techniques and laser irradiation may also be useful, but corticosteroids are ineffective.
Cobb Syndrome (Cutaneomeningospinal Angiomatosus)
Lumbosacral hemangiomas or port-wine stains may be associated with underlying spinal angiomas in Cobb syndrome (cutaneomeningospinal angiomatosus). Neurologic symptoms usually develop in later childhood or adolescence, but early spinal angiography may detect spinal angiomas, which may be surgically excised more easily before they have enlarged.
Maffucci's Syndrome (Hemangiomatosis Osteolytica)
Maffucci's syndrome (hemangiomatosis osteolytica) is a congenital syndrome characterized by hemangiomas at birth, skeletal deformities and enchondromas (benign cartilagenous growths). Malignant degeneration occurs in both the enchondromas and hemangiomas, and suspicious lesions should be biopsied. Diagnosis of this syndrome is confirmed by clinical, radiographic and pathologic evidence of hemangiomas and enchondromas.11 Treatment is symptomatic, and the vascular lesions respond to surgery, irradiation and sclerotherapy. Orthopedic evaluation of bony abnormalities is especially important in the adolescent period.
Treatment
In general, the goals of hemangioma treatment are to prevent loss of life or function, and to prevent scarring, either before or as a result of therapy. The physician should also consider psychosocial issues when deciding on the best course of treatment. Most hemangiomas require no specific therapy other than patient education and reassurance. Frequent follow-up of an infant with a growing hemangioma will ensure adequate treatment of potential sequelae. Spontaneous involution often leads to ulceration; however, permanent scarring occurs in less than 5 percent of children affected with ulceration.
Bleeding and infection also complicate involution and should be treated with direct pressure, wet compresses and antibacterial soap, and oral antibiotics when cellulitis is present.12 Treatment with FPDL and topical antibiotics may hasten recovery. Less than 10 percent of hemangiomas require therapy for specific indications2,3,12 (Table 2). Although the therapeutic management of hemangiomas has included cryotherapy and radiation therapy, these methods have been surplanted by steroids, interferon alfa-2a, laser, surgery, embolization, sclerosing agents and antifibrinolytics.
TABLE 2 Treatment Indications for Hemangiomas
| Threat to life or function | |
| Kasabach-Merritt syndrome (coagulopathy) | |
| Anatomic site | |
| Vision impairment | |
| Respiratory impairment | |
| High-output cardiac failure (mortality up to 50 percent)11 | |
| Hepatic lesions | |
| Other internal lesions | |
| Location in scarprone area | |
| Nose | |
| Lip | |
| Ear | |
| Glabellar area | |
| Any large facial hemangiomas | |
| Pedunculated lesions | |
| Tendency to bleed or to become infected | |
| Rapid rate of growth (tripling in size within weeks) | |
Corticosteroids. The therapeutic benefit of systemic corticosteroids in managing rapidly growing, life-threatening or functionally disabling hemangiomas is well-recognized. Prednisone or prednisolone is administered in a single oral morning dose of 2 to 4 mg per kg per day for two to three weeks, with a response rate of 30 to 90 percent. A positive response, characterized by tactile softening, lightening color or slowed growth, should occur within seven to 10 days of initiation of therapy, and the full dose is then continued for four to six weeks and tapered slowly as the lesion stops growing.12 If rebound growth occurs, an increased dosage of corticosteroid for two weeks is appropriate. If no response is seen within the initial week of treatment, corticosteroids should be discontinued. Systemic side effects of corticosteroid therapy include cushingoid symptoms, growth retardation and infection.
Intralesional corticosteroid therapy may be useful in the management of periorbital hemangiomas. A combination of betamethasone acetate, in a dosage of 6 to 12 mg, and triamcinolone acetonide, in a dosage of 40 to 60 mg, administered with a 30-gauge needle, results in a good response in 45 percent of patients.13 A response occurs within one week; additional injections are required in four to eight weeks. Direct injection in an infant requires sedation. Complications of local injection include eyelid necrosis, cholestin plaque deposits, fatty or soft tissue atrophy and central retinal artery occlusion.14
The exact mechanism of action of corticosteroids in inducing involution of hemangioma is unknown. Corticosteroids inhibit activators of fibrinolysis in vessel walls, decrease plasminogen activators and increase the sensitivity of precapillary sphincters to vasoactive amines. In combination with mast cell–derived heparin, steroids inhibit angiogenesis and induce capillary regression.15
Interferon Alfa-2a. Interferon alfa-2a is the preferred therapy for life- or sight-threatening hemangiomas unresponsive to corticosteroids. Interferons inhibit angiogenesis and stimulate endothelial cell prostacyclin formation, which prevents platelet trapping.16
In a recent study,17 18 of 20 infants whose lesions were resistant to steroid therapy responded to interferon alfa-2a, with a 50 percent regression rate after an average of seven months of therapy. Because of its slow response time, interferon alfa-2a is not effective in the management of acutely obstructing hemangiomas, but it has been described as an effective treatment for Kasabach-Merritt syndrome18 and diffuse neonatal hemangiomatosis. Interferon alfa-2a is administered in daily subcutaneous injections of 1 to 3 million units per square meter of body surface area. Acute side effects, which are reversible, include fever, chills, arthralgias and retinal vasculopathy. Fatigue, leukopenia, anemia, thrombocytopenia, nausea, vomiting, elevated liver transaminase levels and neurotoxicity may occur with chronic use.
Laser Therapy. Several types of lasers are used in the treatment of hemangiomas. FPDL penetrates to a depth of 1.8 mm and has a low risk of scarring. It is the treatment of choice for superficial strawberry hemangiomas and vascular malformations, including port-wine stains and salmon patches, with a response rate of 94 percent for the port-wine stains and salmon patches, and 60 percent for superficial hemangiomas.6,19 This laser also effectively treats residual telangiectasias remaining after spontaneous regression of hemangiomas. Since progression of the deeper portions of a superficial hemangioma cannot reliably be prevented by treatment with FPDL, the neodynium:YAG (Nd:YAG) laser, with a coagulating depth of 5 to 6 mm, is recommended in the treatment of rapidly growing mixed or deep hemangiomas.19 Although 75 percent of patients treated with the Nd:YAG laser exhibit dramatic regression of deep or mixed hemangiomas, scar formation with this laser is more frequent than with the FPDL since this laser penetrates deeper into the skin. Because of the risk of rapid enlargement of some hemangiomas, early treatment is recommended in patients with functionally or cosmetically compromising lesions.
The carbon dioxide laser is effective in treating subglottic hemangiomas unresponsive to corticosteroids and may prevent the need for tracheostomy in patients with airway obstruction. Because of the increased risk for scarring with both carbon dioxide and argon lasers, these lasers are not recommended in the initial treatment of cutaneous hemangiomas. Application of eutectic mixture of local anesthetic (EMLA) cream is effective in reducing the pain associated with FPDL treatment; however, the Nd:YAG, carbon dioxide and argon lasers require local or general anaesthesia.5
Surgery. Surgical excision is occasionally advocated as primary treatment of hemangiomas. Surgical excision is clearly indicated in the management of visceral or ocular lesions unresponsive to corticosteroids, and in the cosmetic revision of redundant skin remaining after spontaneous involution of deeper hemangiomas.12,20 Embolization is a primary treatment for inoperable lesions, and it may be used preoperatively to minimize intraoperative blood loss. Potential side effects include cerebrovascular accident from the backflow of particles into the internal carotid artery.
Antifibrinolytics. Aminocaproic acid (Amicar) and tranexamic acid (Cyclokapron) are antifibrinolytic agents with limited roles in the treatment of Kasabach-Merritt syndrome that is unresponsive to high-dose cortico-steroids or interferon alfa-2a therapy.5 These agents inhibit plasminogen activator and plasmin. Side effects include nausea, vomiting, diarrhea and, in the case of aminocaproic acid, rare instances of renal insufficiency and myopathy.
