While the evaluation and treatment of patients with seizures or epilepsy is often challenging, modern therapy provides many patients with complete seizure control. After a first seizure, evaluation should focus on excluding an underlying neurologic or medical condition, assessing the relative risk of seizure recurrence and determining whether treatment is indicated. Successful management of patients with recurrent seizures begins with the establishment of an accurate diagnosis of epilepsy syndrome followed by treatment using an appropriate medication in a manner that optimizes efficacy. The goal of therapy is to completely control seizures without producing unacceptable medication side effects. Patients who do not achieve complete seizure control should be referred to an epilepsy specialist, since new medications and surgical treatments offer patients unprecedented options in seizure control.
Epileptic seizures are a common and important medical problem, with about one in 11 persons experiencing at least one seizure at some point.1 Epilepsy—the tendency to have recurrent, unprovoked seizures—occurs with a prevalence of about 0.5 percent and a cumulative lifetime incidence of 3 percent.2 The management of patients with epilepsy is often challenging, as evidenced by a recent report that over one half of all patients with epilepsy continue to experience at least occasional seizures despite treatment with antiepileptic medications.3 New clinical advances offer considerable hope to these patients. This article reviews the practical clinical implications of recent studies on the epidemiology, diagnosis and management of epilepsy.
Management of Patients with New Seizures
Despite numerous technologic advances in the evaluation of neurologic disorders, diagnosis of the first seizure is still based predominantly on the patient's medical history. Many paroxysmal events may be confused with epileptic seizures, including syncope, movement disorders, parasomnias and psychogenic seizures.
Probably the most common entity that is confused with epileptic seizures is syncope. Studies in which volunteers were videotaped during induced syncopal events illustrate the common occurrence of repetitive clonic, myoclonic or dystonic movements on fainting.4 These movements, however, rarely persist beyond five to 10 seconds and do not exhibit the organized progression from tonic to clonic phase seen in a convulsive seizure. Thus, a detailed history of the motor activity, together with the usual questions regarding premonitory symptoms, postictal state, tongue-biting, incontinence and provoking factors, can often help distinguish between these two common entities.
Diagnostic Evaluation
Diagnostic studies must be tailored to individual patients. Basic laboratory evaluation focuses on detecting systemic disturbances potentially associated with seizures and includes a complete blood count and measurements of electrolytes, calcium, magnesium, phosphorus, blood urea nitrogen, creatinine and glucose. Consideration also should be given to obtaining a toxicology screen and evaluating hepatic function with synthetic and enzyme studies. Lumbar puncture is essential in patients in whom meningitis or encephalitis is suspected, as well as in immunocompromised patients, since occult meningitis is a common finding in this group.5
Most authorities recommend that all patients who experience an unprovoked seizure undergo a brain imaging study in an effort to detect underlying cerebral lesions (e.g., tumor, abscess, vascular malformation, stroke, traumatic injury). In nonurgent cases, the imaging modality of choice is magnetic resonance imaging (MRI), since it is more sensitive than computed tomography (CT) in identifying these lesions. In patients presenting with a seizure in whom the history or examination suggests new focal deficits, persistent altered mental status, fever, recent trauma, persistent headache, cancer, treatment with anticoagulation or immunocompromised state, emergent neuroimaging is recommended.6 This is usually accomplished with a CT scan, given its widespread availability and speed and its superior ability in the detection of acute hemorrhage, compared with MRI.
Electroencephalography (EEG) is often helpful in the evaluation of patients presenting with a seizure. The utility of EEG includes detection of epileptiform activity, strengthening the putative diagnosis; identification of focal electrocerebral abnormalities suggesting a focal structural brain lesion; and documentation of specific epileptiform patterns associated with particular epilepsy syndromes (for example, generalized spike-and-wave discharges associated with a generalized epilepsy, or focal discharges associated with a localization-related epilepsy).
Multiple recordings increase the diagnostic yield of the EEG; while approximately 30 to 50 percent of patients with epilepsy demonstrate epileptiform abnormalities on the first EEG, the yield increases to 60 to 90 percent with repeated recordings.7 Various maneuvers increase the likelihood of recording epileptiform discharges, including sleep deprivation before the study, recording a portion of the EEG as the patient sleeps, recording the EEG as the patient hyperventilates for several minutes, and subjecting the patient to stroboscopic photic stimulation during the study. The EEG is also helpful in assessing the risk of seizure recurrence. A recent prospective study of EEGs performed in patients with untreated idiopathic first seizures found that the presence of epileptiform abnormalities on EEG was associated with a recurrence risk of 83 percent, compared with 41 percent in patients with nonspecific abnormalities and 12 percent in patients in whom routine and partially sleep-deprived EEGs were normal.8
While most physicians prefer not to prescribe ongoing antiepileptic therapy for patients with a single seizure, the decision to treat initial seizures with medication remains controversial. Several factors should be considered when making a decision, including the likelihood of recurrent seizures, the risk of the treatment itself, the ability of the treatment to decrease the risk of recurrent seizures and the consequence of further seizures to the patient. Since these factors vary from patient to patient, it seems unlikely that a single approach can be recommended. Recent studies have better defined some of these factors so that treatment decisions can be individualized.
Early nonrandomized studies of antiepileptic drug treatment in patients presenting with a first seizure did not demonstrate a reduced likelihood of seizure recurrence. These data were used to support the concept of not treating a patient with a first seizure, although the inherent bias associated with nonrandomization was probably responsible for the results. In three recent randomized trials, antiepileptic drug treatment was associated with a significant reduction in recurrence risk, although the benefit persisted only for the time that the patient was taking medication.9–11 Thus, taking an antiepileptic drug for a discrete period of time did not decrease the long-term risk of eventually developing a chronic seizure disorder.12 The finding that antiepileptic drugs are effective in preventing recurrent seizures is important in that patients with a very high risk of seizure recurrence may choose to take medication to lessen that risk.
In a meta-analysis13 of 16 studies with median follow-up ranging from one to five years, the risk of seizure recurrence following an unprovoked seizure was 51 percent. The two most important prognostic factors influencing risk of recurrence included etiology of the seizures and EEG findings. Patients whose seizures were likely symptomatic of remote cerebral injury were more likely to experience recurrence than those whose seizures were judged to be idiopathic, with two-year recurrence risks of 57 percent versus 32 percent.13 As previously mentioned, patients with epileptiform disturbances (spikes or sharp waves) on the interictal EEG are more likely to experience recurrence than those with non-epileptiform abnormalities or those with a normal EEG.8 This information helps to individualize treatment and counseling after a patient's first seizure.
In addition to considering the probability of recurrence, one must also consider the potential psychologic, social and vocational consequences of further seizures. Since children are not as likely to have severe social or vocational repercussions from a single recurrence, they are rarely treated for a first seizure. Some adults, however, feel that the consequences of even a single recurrent seizure would dramatically affect their lives, and they may choose to take medication to decrease the chance of a recurrence.
Medical Management of Epilepsy: General Principles
The goal of treating patients with epilepsy is to control seizures completely without causing unacceptable side effects. In the past several years, a number of new antiepileptic drugs have become available, and more will soon be released.
To achieve optimal treatment results, several strategies should be used (Table 1). The most important step is to select an antiepileptic drug that is appropriate for the patient's particular type of epilepsy. A specific epilepsy syndrome diagnosis is based on the history of the patient's seizure types, neurologic status and EEG findings (Table 2).
TABLE 1 Medication Treatment Strategies for Patients with Epilepsy
| Establish an epilepsy syndrome diagnosis for each patient (Table 2) |
| Select medications appropriate for that epilepsy syndrome (Table 2) |
| From the appropriate medications, choose the agent best suited for the patient based on patient and medication characteristics (Table 3) |
| Initiate and titrate the medication at appropriate dosages, increments and rates to enhance tolerability (Table 3) |
| Increase the medication, regardless of serum levels, until complete seizure control is achieved or until persistent, unacceptable side effects occur |
| If satisfactory seizure control is not achieved, change to another agent appropriate for the epilepsy syndrome being treated; the goal should be antiepileptic drug monotherapy in each patient, when possible |
| If trials with one or two agents fail to achieve acceptable results, refer the patient to an epilepsy specialist for consultation |
TABLE 2 Epilepsy Syndromes
| Generalized* | Localization-related (partial or focal)† | |||
|---|---|---|---|---|
| Idiopathic‡ | Seizure types: absence, myoclonic, tonic-clonic | Seizure types: simple partial (awareness unimpaired), complex partial (awareness impaired), secondarily generalized tonic-clonic | ||
| Neurologic examination: normal | Neurologic examination: normal | |||
| Neuroimaging: normal | Neuroimaging: normal | |||
| EEG: Normal background with fast (3 to 6 Hz) generalized spike-and-wave discharges | EEG: Normal background with focal epileptiform discharges | |||
| Common examples: | Common examples: | |||
| Childhood absence epilepsy Juvenile myoclonic epilepsy Epilepsy with generalized tonic-clonic seizures on awakening | Benign childhood epilepsy with centro-temporal spikes (Rolandic epilepsy) Benign epilepsy with occipital paroxysms | |||
| Treatment: valproate (Depakote), ethosuximide (Zarontin [effective for absence seizures only]), lamotrigine (Lamictal), topiramate (Topamax), felbamate (Felbatol) | Treatment: carbamazepine (Tegretol), phenytoin (Dilantin) | |||
| Symptomatic¶ | Seizure types: atypical absence, myoclonic, tonic, atonic, tonic-clonic | Seizure types: simple partial (awareness unimpaired), complex partial (awareness impaired), secondarily generalized tonic-clonic | ||
| Neurologic examination: diffuse or multifocal abnormalities | Neurologic examination: focal abnormalities or normal | |||
| Neuroimaging: diffuse or multifocal abnormalities common | Neuroimaging: focal abnormalities common | |||
| EEG: Abnormal background with slow (<3 Hz) generalized and/or multifocal epileptiform discharges | EEG: Normal or abnormal background with focal or multifocal epileptiform discharges | |||
| Common examples: | Common examples: | |||
| Lennox-Gastaut syndrome | Temporal lobe epilepsy | |||
| Progressive myoclonus epilepsies | Frontal lobe epilepsy | |||
| Treatment: valproate, lamotrigine, topiramate, felbamate, ketogenic diet, corpus callosotomy | Treatment: carbamazepine, phenytoin, valproate, new agents (gabapentin [Neurontin], lamotrigine, tiagabine [Gabitril] topiramate, felbamate), vagus nerve stimulator, resective surgery | |||
EEG = electroencephalogram.
*—Generalized epilepsies: seizures begin diffusely throughout cerebral cortex.
†—Localization-related epilepsies: seizures arise from a discrete focus in cerebral cortex or limbic structures (hippocampus or amygdala).
‡—Idiopathic epilepsies are primary in nature, without clear cause.
¶—Symptomatic epilepsies are secondary in nature, resulting from an apparent or assumed brain lesion.
The four broad categories of epilepsy syndromes are idiopathic generalized, symptomatic generalized, idiopathic localization-related and symptomatic localization-related. The importance of making a syndromic diagnosis is illustrated by juvenile myoclonic epilepsy, a common idiopathic generalized epilepsy syndrome in which patients may experience absence, myoclonic and convulsive (clonic-tonic-clonic or tonic-clonic) seizures. If phenytoin (Dilantin) or carbamazepine (Tegretol) is used for treatment, the nonconvulsive seizures are often worsened. In contrast, valproate (Depakote) provides complete seizure control in most patients.
While patients with generalized-onset epilepsies (such as juvenile myoclonic epilepsy) respond best to valproate, any of the major medications (except ethosuximide [Zarontin]) may be effective in patients with localization-related (partial-onset) epilepsy. In patients with partial epilepsy, large studies have consistently demonstrated similar efficacy for phenytoin, carbamazepine, valproate, primidone (Mysoline) and phenobarbital, although the barbiturates are often poorly tolerated as a result of their sedating properties.14–16
Thus, in patients with localization-related epilepsies, drug selection is heavily influenced by the side effect profile, cost and dosing frequency of each agent (Table 3). The long-term cosmetic consequences (which include coarsening of the facial features, gingival hyperplasia, hirsutism and enlargement of the lips) for many patients taking chronic phenytoin therapy, for example, make this a poor first choice for children and young adults. Yet, the once-daily dosing scheme and low cost make it an attractive agent for other patients. Carbamazepine, especially the new extended-release preparations, is an excellent choice for many children and adults with localization-related epilepsies. Valproate, in addition to being the drug of choice for most of the generalized epilepsies, is also efficacious in the treatment of the localization-related syndromes. Accordingly, it is a good first choice in patients in whom the epilepsy syndrome is not clearly defined.
Once the preferred medication has been chosen, therapy should be initiated at an appropriate dosing schedule and increased at an appropriate dosing increment and rate (Table 3). For any of the antiepileptic drugs, we recommend that dosage increases continue—regardless of serum drug levels—until complete seizure control is achieved or until persistent, unacceptable side effects occur.
TABLE 3 Oral Antiepileptic Medications
| Generic name | Trade name | Strengths available* (mg) | Typical adult starting dosage† | Typical increment and rate of ascension‡ | Most common dose-related adverse effects | Non–dose-related and idiosyncratic reactions | Cost per month¶ |
|---|---|---|---|---|---|---|---|
| Carbamazepine | Tegretol | 100, 200 | 200 mg twice a day | 200 mg per week (taken three or four times a day) | Dizziness, somnolence, ataxia, nausea, vomiting, diplopia, blurred vision | Hyponatremia, rash, Stevens-Johnson syndrome, leukopenia, aplastic anemia, agranulocytosis, transaminitis, hepatic failure | $ 68.00 (1,200 mg per day) |
| Tegretol-XR | 100, 200, 400 | 200 mg twice a day | 200 mg per week (taken twice a day) | 68.00 | |||
| Carbatrol | 200, 300 | 200 mg twice a day | 200 mg per week (taken twice a day) | N/A | |||
| Ethosuximide | Zarontin | 250 | 250 mg every day to 250 mg twice a day | 250 mg per week | Anorexia, nausea, vomiting, drowsiness, headache, dizziness | Rash, Stevens-Johnson syndrome, hemopoietic complications | 71.00 (750 mg per day) |
| Felbamate | Felbatol | 400, 600 | 400 mg three times daily | 400 to 600 mg per week | Anorexia, vomiting, insomnia, nausea, headache, dizziness | Aplastic anemia, hepatic failure | 174.00 (3,600 mg Per day) |
| Gabapentin | Neurontin | 100, 300, 400 | 300 mg daily to three times a day | 300 mg per week (taken three or four times a day) | Somnolence, dizziness, ataxia, fatigue | Rash, weight gain, behavior changes, peripheral edema | 240.00 (2,700 mg Per day) |
| Lamotrigine | Lamictal | 25, 100, 150, 200 | 25 mg every other day (with valproate [Depakote]), to 25 mg twice a day (with carbamazepine, phenobarbital or phenytoin [Dilantin]) | 25 mg per 2 weeks (taken twice a day) | Dizziness, ataxia, somnolence, headache, diplopia, blurred vision, nausea, vomiting, rash | Rash, Stevens- Johnson syndrome, transaminitis | 174.00 (600 mg per day |
| Phenobarbital | 15, 30, 60, 100 | 100 mg every day | 15 to 30 mg per week | Somnolence, cognitive and behavior effects | Rash, Stevens-Johnson syndrome, hemopoietic complications, transaminitis, hepatic failure | 4.00 (200 mg per day) | |
| Phenytoin | Dilantin | 30, 50, 100 | 300 mg every day | 25 to 30 mg per week | Ataxia, diplopia, slurred speech, confusion | Rash, Stevens-Johnson syndrome, hemopoietic complications, gingival hyperplasia, coarsening of facial features, transaminitis, hepatic failure | $ 32.00 (400 mg per day) |
| Tiagabine | Gabitril | 4, 12, 16, 20 | 4 mg every day | 4 mg per week (taken two to four times a day) | Dizziness, nervousness, asthenia, confusion, tremor | Not established | 149.00 (56 mg per day) |
| Topiramate | Topamax | 25, 100, 200 | 25 mg twice a day | 50 mg per week | Somnolence, dizziness, ataxia, slurred speech, psychomotor slowing, cognitive problems | Anemia, acne, alopecia, weight loss, transaminitis, nephrolithiasis | 173.00 (400 mg per day) |
| Valproate | Depakote | 125, 250, 500 | 250 mg three times a day | 250 mg per week | Nausea, vomiting, tremor, thrombo-cytopenia | Weight gain, hair changes/loss, transaminitis, hepatic failure, rash, Stevens-Johnson syndrome | 106.00 (1,500 mg per day) |
*—Strengths listed are for tablet or capsule formulations of the brand name agents.
†—Initiation dosages for some agents vary, depending on concomitant medications, body weight, age of patient and other factors; consult prescribing information for each drug. Dosages are for nonurgent initiation of medication; clinical circumstances may necessitate increased dosages and accelerated titration. See prescribing information for pediatric dosages, which are based on body weight and often must be administered more frequently than in adult dosages.
‡—Rate of increase may need modification, depending on seizure frequency and occurrence of adverse effects. Note that phenytoin may be increased in 25-mg increments by using a halved 50-mg Dilantin Infatab tablet or by 30 mg using a 30-mg Dilantin Kapseal capsule.
¶—Estimated cost to the pharmacist for typical adult maintenance dosages based on average wholesale prices in Red book. Montvale, N.J.: Medical Economics Data, 1997. Cost to the patient will be higher, depending on prescription filling fee.
For patients with relatively infrequent seizures (in whom it is difficult to gauge the response to treatment without waiting many months or years for the next seizure to occur), we believe a logical approach is to promptly increase the medication to the maximum tolerated dosage and maintain it at this level. This can be accomplished by increasing the agent until the patient begins to experience expected dose-related side effects, and then reducing the dosage to the immediately previous dosage that did not produce the adverse effects. Once a steady state with the refined dosage has been achieved, it is useful to check the trough drug serum level as a reference point for the maximum tolerated dosage.
If the patient eventually experiences a seizure when the serum level is at the reference point, the trial of medication can be considered a failure. This procedure enables assessment of efficacy in a manner significantly more efficient than simply beginning at an average dosage and waiting for the next seizure before the next increase is implemented. If adequate seizure control—which should be defined as complete seizure control for most patients—is not achieved at the maximum tolerated dosage of the first medication, consideration should be given to referring the patient for neurologic consultation, if this has not yet been undertaken. Usually, a second agent will need to be added.
As the dosage of the new medication is titrated up, the original medication is gradually tapered until monotherapy with the new agent is achieved. The particulars of the transition are dictated by the specific pharmacokinetic and pharmacodynamic interactions of the medications being used and the clinical status of the patient. The new medication is then increased to the maximum tolerated dosage. We recommend that patients who fail to respond to two medications be promptly referred to an epileptologist for further evaluation.
A common error in the use of antiepileptic drugs is to base dosing on serum levels. Serum levels provide only a rough indication of the likelihood of response or dose-related side effects. In order to achieve complete seizure control, many patients require serum levels above the upper “therapeutic” limit (“toxic” levels) but tolerate these levels without ill effects. Furthermore, toxic levels reflect the serum concentration at which patients may experience dose-related side effects, symptoms that may be bothersome but are generally not life-threatening and are fully reversible with reduction of the dosage. Thus, toxic antiepileptic drug levels are unlike toxic levels for other drugs (e.g., digoxin, lithium, theophylline), in which elevated levels are often associated with serious medical complications and are, therefore, to be avoided. In the treatment of patients with epilepsy, the clinical status of the patient—seizure control and occurrence of adverse effects—should guide dosing of antiepileptic drugs, rather than serum drug levels.
Serum drug levels are useful for documenting the level corresponding to the maximum tolerated dosage of antiepileptic medication, assessing medication status and patient compliance when a breakthrough seizure has occurred, sorting out the probable cause of nonspecific medication side effects when patients are taking multiple medications, ensuring an appropriate medication level in patients who are unable to report adverse effects (for example, young children and cognitively impaired individuals), and titrating medication dosages throughout pregnancy. In the absence of one of these clinical indications, repeated “routine” determination of antiepileptic drug levels are of little value.
In general, obtaining other laboratory studies on an ongoing, “routine” basis is also of little value.17 We recommend obtaining hematologic and serum chemistry studies (complete blood cell count with differential and platelet count, electrolytes and liver enzymes) before instituting any antiepileptic medication to establish a baseline and to identify any preexisting abnormalities. We find that repeat studies during the early phases of treatment (for example, at one and three months) are sometimes useful in identifying abnormalities that may be reflected in laboratory studies but are not yet apparent clinically (for example, significant thrombocytopenia or hyponatremia). Laboratory studies should also be obtained if a patient presents with signs or symptoms compatible with a possible drug-induced condition and on a routine (perhaps annual) basis in patients less able to communicate untoward effects (such as multiply handicapped institutionalized patients). Patients being treated with felbamate (Felbatol) require more frequent laboratory monitoring. Specific recommendations are provided in the package insert for the drug.
Medical Management: The New Agents
Before the release of felbamate in 1993, no new antiepileptic drug had been licensed in the United States for 15 years. Over the past four years, five new drugs have been introduced, and five new formulations of previously available drugs have been released. These new agents offer physicians expanded treatment options for patients, although the exact roles for these medications are still being determined.18
Felbamate was approved by the U.S. Food and Drug Administration (FDA) in 1993 for use as adjunctive (i.e., add-on) treatment or monotherapy in adults with localization-related epilepsy and as adjunctive therapy in children with Lennox-Gastaut syndrome (symptomatic generalized epilepsy consisting of multiple types of generalized seizures). Unfortunately, felbamate has been associated with two types of often fatal idiosyncratic reactions, aplastic anemia and fulminant hepatic failure, and therefore should only be used in patients in whom the benefits clearly outweigh the risks. Ideally, patients should be evaluated and managed by an epileptologist if the use of felbamate is under consideration.
Gabapentin (Neurontin), licensed in the United States in late 1993, is approved for use as adjunctive therapy in patients 12 years of age or older with localization-related epilepsy. Gabapentin offers a number of unique pharmacokinetic and pharmacodynamic properties, making it well suited for use in patients in whom drug-drug interactions must be avoided (such as those with multiple medical problems, those taking other medications and the elderly). A disadvantage of the agent is the requirement for three or four times daily dosing.
Lamotrigine (Lamictal) was approved in 1994 as adjunctive treatment for localization-related epilepsy in adults. It appears to exhibit a broad spectrum of antiepileptic activity. The agent is used as an alternative in patients with localization-related or generalized epilepsies. Evidence from controlled studies19 and clinical use suggests that lamotrigine is particularly helpful in patients with symptomatic generalized epilepsies. It is typically taken twice daily. In April 1997, a new boxed warning was added to the labeling of lamotrigine concerning reports of severe, potentially life-threatening rashes, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Rates for potentially serious rashes, estimated from clinical studies, are approximately one in 1,000 for adults and one in 50 to 100 in children. As use of this agent increases, a more accurate determination of the frequency of life-threatening rash should be possible. In the United States, lamotrigine is not licensed for use in children and, at the present time, we recommend its use only in children with epilepsy refractory to other medications in whom the potential benefits of the drug outweigh the risks and preferably under the supervision of an epilepsy specialist.
Topiramate (Topamax), released in early 1997, is indicated for use as adjunctive therapy in adults with localization-related epilepsy. Preliminary studies also suggest efficacy in some generalized epilepsies. Cognitive effects constitute the main dose-limiting toxicity for topiramate, and patients taking this drug have an increased incidence of nephrolithiasis.
Tiagabine (Gabitril) received FDA approval in October 1997 as adjunctive therapy for patients 12 years of age or older with localization-related epilepsy. This novel antiepileptic drug selectively inhibits uptake of gamma-aminobutyric acid (GABA) and prolongs the duration of inhibitory activity at postsynaptic receptors. Initial studies suggest that dose-limiting side effects are mainly neurocognitive in nature, such as dizziness and nervousness.
Fosphenytoin (Cerebyx), a parenteral phenytoin prodrug, provides several significant advantages over standard parenteral phenytoin. It is easier to administer, is better tolerated and can be administered intravenously or intramuscularly. Uses for the agent include intravenous administration for the treatment of status epilepticus and treatment of patients who are unable to take oral medication or in whom a more rapid attainment of a therapeutic drug level is desired. The only disadvantage of the agent is its increased expense compared with standard parenteral phenytoin.
Extended-release carbamazepine (Tegretol-XR and Carbatrol) allows twice-daily dosing. These formulations provide increased convenience to patients, improve patient compliance and provide more consistent serum levels of carbamazepine (potentially reducing peak-level toxicity and trough-level seizures). Tegretol-XR consists of an extended-release tablet with a unique delivery system that must not be divided or chewed. The tablet is not digested, although its contents are slowly released during gastrointestinal transit. Carbatrol is formulated as a multi-component capsule consisting of immediate-, extended-and enteric-release beads. The capsule may be swallowed intact or opened and sprinkled on food for use in young children.
A parenteral preparation of valproate (Depacon) is now available and is indicated as an intravenous alternative in patients for whom oral administration of valproate is temporarily not feasible. At present, the agent is not indicated for the treatment of status epilepticus.
Diazepam rectal gel (Diastat) recently became available in the United States and is helpful in controlling acute repetitive seizures in patients with refractory epilepsy. It is indicated for intermittent use in the management of selected, refractory patients with epilepsy on stable antiepileptic drug regimens who are prone to bouts of increased seizure activity. The main benefit of the formulation is the ease of rectal administration.
The Medically Refractory Patient
Participants in a 1990 National Institutes of Health consensus conference estimated that approximately 20 percent of patients with epilepsy, or 400,000 Americans, have intractable seizures.20 The arrival of newly licensed antiepileptic drugs is unlikely to dramatically alter the number of patients with refractory epilepsy. One review of experimental trials suggests that less than 2 percent of patients in whom conventional medications have failed will become seizure-free during a trial with a new medication.21 Since the possibility of obtaining seizure control diminishes with successive drug trials, we recommend that patients be referred to a comprehensive epilepsy center if two or three first-line medications fail to control seizures.
The problem of refractory seizures is important since patients with ongoing seizures are subject to considerable physical risk. Studies consistently suggest that people with epilepsy, especially those who experience convulsions, are at risk of dying as a consequence of their seizures. Indeed, the lifetime risk of dying a seizure-related death (from status epilepticus, accidents or sudden unexplained death) is approximately 25 percent in patients with poorly controlled epilepsy.22 When seizures can be controlled for an extended period, however, rates of mortality approach those of the general population.23
The psychosocial consequences of poorly controlled seizures are also of major concern. Patients with epilepsy are generally undereducated and underemployed for their level of function. Recently, investigators have found that employment status improves in patients following seizure surgery, especially if seizures are abolished.24 Even rare residual seizures adversely affect the chance for employment. These observations suggest that complete seizure control may be necessary to optimize a patient's quality of life. In addition, the psychosocial and physical consequences of imperfectly controlled epilepsy underscore the importance of prompt referral to a center specializing in the diagnosis and care of patients with epilepsy when patients fail to completely respond to treatment.
Several options exist for patients whose seizures prove refractory to the licensed antiepileptic drugs, including treatment with experimental medications as part of an investigational protocol, the ketogenic diet, implantation of a vagus nerve stimulator and seizure surgery.
Vagus Nerve Stimulator
The vagus nerve stimulator is a novel, non-pharmacologic treatment for epileptic patients whose seizures are uncontrolled by medication. The device was approved by the FDA in mid-1997 for implantation in patients over 12 years of age with medically refractory localization-related epilepsy. Under general anesthesia, a bipolar lead is wrapped around the left vagus nerve and tunneled to the infraclavicular region, where it is connected to a signal generator. This signal generator delivers a precise pattern of stimulation to the vagus nerve; the parameters for stimulation are non-invasively programmed by the physician using a computerized programming wand. Typically, the device stimulates for 30 seconds every five minutes. In addition, by using a hand-held magnet the patient or a caregiver can manually activate the stimulator at the onset of a seizure with the goal of terminating the seizure before it escalates. Randomized, controlled and blinded studies have demonstrated the vagus nerve stimulator to be efficacious in reducing the frequency of seizures in some patients, with approximately 30 percent of patients experiencing at least a 50 percent reduction in seizure frequency.25,26 Ongoing trials are evaluating the effectiveness of the device in reducing seizures associated with the generalized epilepsies.
Seizure Surgery
Seizure surgery should be considered for patients in whom antiepileptic drugs fail to completely control seizures. In determining whether a patient with intractable epilepsy is a surgical candidate, it is important to confirm an anatomic and syndromic diagnosis appropriate for surgical treatment (Table 2). Patients with progressive metabolic or neurodegenerative conditions are generally considered poor candidates for surgery. Otherwise, most patients with intractable epilepsy should be considered potential candidates for seizure surgery.
Anterior temporal lobe resection is the most common procedure performed for the treatment of epilepsy. Since surgery often results in complete seizure control, studies of epilepsy surgery differ from medication trials in that they typically recognize complete control of seizures as a primary outcome measure. Since seizure control is the most important goal of surgery, the operation is considered a success if the patient becomes seizure-free, even if medication is still required.
Over time, the trend has gradually turned toward better surgical outcome in patients undergoing anterior temporal resections.27 Improved outcomes probably reflect the enhanced ability to localize epileptogenic tissue with modern imaging and EEG techniques (Figure 1). Currently, 80 percent of all patients undergoing temporal lobe resections at our center become seizure-free. Furthermore, patients with evidence of mesial temporal sclerosis on MRI have a prospect for a seizure-free outcome that is greater than 90 percent.28
FIGURE 1.
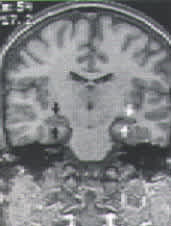
High-resolution coronal magnetic resonance image in a patient with longstanding medically refractory temporal lobe epilepsy. Atrophy of the left hippocampus is apparent (light arrows). The right hippocampus is of normal size and appearance (dark arrows). Patients with the finding of unilateral hippocampal atrophy and ictal electroencephalographic recordings that demonstrate concordant seizure localization have a very high likelihood (greater than 90 percent) of becoming seizure-free with surgical treatment.
Occupational outcome improves after surgery in patients of all ages, although patients who undergo surgery earlier typically achieve a better occupational outcome score as a result of better preoperative function.29 Since patients with chronic epilepsy often have been chronically disabled from their vocation, it is not surprising that they do not become employed immediately after successful surgery.24
Most patients undergoing seizure surgery are young and otherwise healthy, which probably explains the low rates of surgical morbidity. Permanent, severe deficits related to the surgery are rare. However, subtle changes in verbal skills, especially naming, are sometimes present postoperatively in a minority of patients undergoing language-dominant temporal lobe resections.30
Final Comment
Patients with epilepsy now have available to them more therapeutic options than ever before. In order for patients to benefit from these advances, physicians must be attuned to making an accurate diagnosis of epilepsy syndrome, selecting and using medications properly, and promptly referring patients who do not completely respond to treatment to a comprehensive epilepsy center.
