Cutaneous T-cell lymphoma, also known as mycosis fungoides, is a malignancy of the T helper (CD4+) cells. Diagnosis is difficult early in the course of this disease because it mimics several benign skin disorders, including eczema, psoriasis and contact dermatitis. Cutaneous T-cell lymphoma is also difficult to identify histologically, and multiple biopsies may be necessary to confirm the diagnosis. Treatment may require a combination of topical and systemic agents. Patients with limited skin disease have a good prognosis, but the prognosis is less hopeful in those with extracutaneous involvement. As the disease progresses, the normal T-cell population is eliminated, and the patient becomes significantly immunosuppressed. Infection is the primary cause of mortality in patients with cutaneous T-cell lymphoma.
Cutaneous T-cell lymphoma (CTCL), also known as mycosis fungoides, is a malignancy of the T-helper (CD4+) cells. It may mimic many benign processes, such as eczema, psoriasis and contact dermatitis. Early in the course of the disease, the clinical and histologic diagnosis of CTCL is difficult. The diagnosis may be missed and the patient left untreated for years because of the benign appearance of this disorder. Close follow-up with multiple biopsies over time may help the physician make the diagnosis. Once the disease becomes systemic, the prognosis is significantly worse. A variety of treatments for CTCL exist, with overall good patient response when treatment is started early.
CTCL can present clinically as patches, plaques, tumors or generalized erythroderma. To further complicate matters, in the early stages, lesions may diminish in intensity if treated with topical corticosteroids, supporting an incorrect diagnosis of a benign process. Although the patient and physician may be reassured by this seemingly well-controlled dermatitis, the correct diagnosis is delayed by topical corticosteroid treatment.1
CTCL is the most common primary cutaneous lymphoma and should be considered if a chronic psoriasiform or eczematous dermatitis has not responded appropriately to treatment. Even though the prognosis is good in most patients with patch-stage disease, extracutaneous spread involving any organ is possible and may eventually lead to death. Definitive diagnosis and treatment of cutaneous lymphoma may slow disease progression.
Clinical Presentation and Differential Diagnosis
HISTORY
Most cases of CTCL are diagnosed in patients 50 to 60 years of age. It is twice as common in black persons as in white persons, but CTCL may affect persons in any age or ethnic group.2 A recent series3 noted that between 4 and 5 percent of patients with CTCL had the disease by 20 years of age. Patients may give a history of pruritic, inflammatory patches or plaques present for several years, even decades. The disease tends to begin as patches on the trunk, which can remain for years before the plaque stage presents. The rash of CTCL frequently resembles psoriasis. Like psoriasis, it may improve with sun exposure. Eventually, tumors or erythroderma may occur as malignant cells proliferate.
PHYSICAL EXAMINATION
Various clinical patterns may be noted in patients with CTCL that reflect the progression of this disease from patch to plaque to tumor. Opinions differ about whether certain skin disorders, such as poikiloderma vasculare atrophicans (PVA), large-plaque parapsoriasis (LPP) and small-plaque parapsoriasis (SPP), represent early presentations of CTCL or are premalignant conditions.4 There is an increasing tendency to regard such abnormal patch-plaque diseases as early forms of CTCL. Early CTCL often lacks the histologic criteria necessary for a definitive diagnosis.5
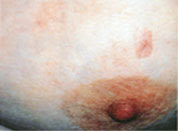
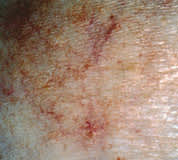
PVA presents as atrophic, hypopigmented and hyperpigmented macules and patches with telangiectasias and tends to be located on the breasts, buttocks and flexured areas (Figure 1). The atrophic skin has a characteristic wrinkled, “cigarette-paper” appearance (Figure 2). Patients with LPP, often considered to be a variation of patch stage CTCL, present with atrophic, inflammatory lesions, primarily over the buttocks and intertriginous areas. Early patch stage CTCL presents as one or more annular scaly macules and patches that are orange or dusky red and often occur on unexposed sites (Figures 3 through 5). After months or years, the patches can evolve into plaques that become scaly and indurated (Figure 6).
FIGURE 1.
Atrophic patch with increased telangiectasias characteristic of cutaneous T-cell lymphoma variant of poikiloderma vasculare atrophicans (PVA).
FIGURE 2.
Poikiloderma vasculare atrophicans (PVA) on the breast, with characteristic “cigarette paper” atrophic appearance.
FIGURE 3.
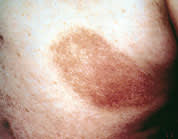
Early patch phase of cutaneous T-cell lymphoma on the back, with dusky red appearance similar to that of an eczematous lesion.
FIGURE 4.
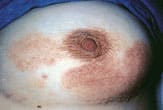
Patch stage of cutaneous T-cell lymphoma mimicking an eczematous rash or drug eruption on the breast.
FIGURE 5.
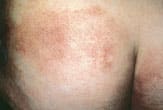
Erythematous patches of cutaneous T-cell lymphoma on the buttocks. This patient was initially thought to have an irritant dermatitis.
FIGURE 6.
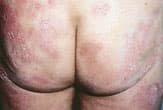
Multiple scaling plaques of cutaneous T-cell lymphoma resembling a psoriatic rash on the buttocks.
If disease progression continues, the tumor stage is usually next. Red to violaceous nodules form and may ulcerate6 (Figure 7). Extracutaneous spread may occur through the regional lymphatic system to the viscera. Sézary syndrome is a systemic variant of CTCL and manifests as erythroderma with generalized bright red, scaling skin and associated leukemia and lymphadenopathy (Figure 8). The erythroderma of preexisting skin diseases such as psoriasis or atopic dermatitis and drug eruption may be considered in the differential diagnosis. Symptoms of weight loss and malaise often may accompany the erythrodermal stage of CTCL.1,5
FIGURE 7.
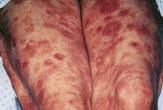
Violaceous nodules apparent on the lower extremities as cutaneous T-cell lymphoma progresses.
FIGURE 8.
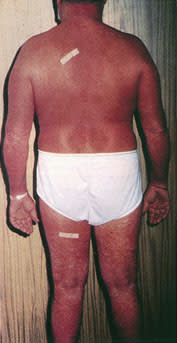
The generalized erythroderma and scaling skin of Sézary syndrome, resembling the erythroderma of drug eruptions and benign skin conditions such as psoriasis and atopic dermatitis.
LABORATORY AND HISTOLOGIC STUDIES
In addition to a thorough history and physical examination, including examination of the lymph nodes, suggested baseline studies include a complete blood count, liver function tests and a radiograph of the chest. If advanced disease is suspected, additional studies, such as computed tomography and bone marrow biopsy, may also be considered. Once peripheral blood and lymph nodes are involved, visceral disease is often present. CTCL can involve virtually any organ.2,5
The diagnosis of CTCL is confirmed by skin biopsy. Several biopsies may be needed over time to make the diagnosis, since the early patch stage may mimic chronic inflammatory dermatoses. Immunophenotyping and T-cell receptor gene arrangement analysis confirming a malignant clone are sometimes helpful in diagnosis.2,7
Prognosis
The presence of extracutaneous disease and the type and amount of skin involvement are the most important prognostic factors in patients with CTCL. A modified staging system to further classify patients was proposed by the International Consensus Conference on CTCL in 1994 (Table 1).8
TABLE 1 Modified Staging System for Cutaneous T-Cell Lymphoma
| Stage | Elements of stage | Prognosis |
|---|---|---|
| IA | T1, LN0-2 | Low risk |
| No adenopathy; no visceral involvement | ||
| IB | T2, LN0-2 | Low risk |
| No adenopathy; no visceral involvement | ||
| IIA | T1-2, LN0-2 | Low risk |
| Palpable lymph nodes; no visceral involvement | ||
| IIB | T3, LN0-2 | Intermediate risk |
| Absent or present palpable lymph nodes; no visceral involvement | ||
| III | T4, LN0-2 | Intermediate risk |
| Absent or present palpable lymph nodes; no visceral involvement | ||
| IVA | T1-4, LN3-4 | High risk |
| Absent or present palpable lymph nodes; no visceral involvement | ||
| IVB | T1-4, LN3-4 | High risk |
| Palpable lymph nodes; visceral involvement |
T1 = plaque <10% body surface area; T2 = plaque >10% body surface area; T3 = one or more cutaneous tumors; T4 = erythroderma; LN0 = lymph nodes uninvolved; LN1 = reactive node; LN2 = dermatopathic node; LN3 = large clusters of convoluted cells; LN4 = obliteration of lymph node.
Adapted with permision from Demierre MF, Foss FM, Koh H. Proceedings of the International Consensus Conference on Cutaneous T-cell Lymphoma (CTCL) Treatment Recommendations. J Am Acad Dermatol 1997;36:460–6.
Patients who have only limited skin disease without extracutaneous disease have a good prognosis, while those with extracutaneous disease have a much poorer prognosis. A study9 of survival data obtained from Stanford University School of Medicine showed that patients with T1 disease who were treated had a median survival of 32.5 years. Those with T2, T3 and T4 disease had survival rates of 12.1 years, 2.9 years and 3.6 years, respectively. Patients with T1 disease are at risk of progression to higher stages. Treatment may prevent or retard disease progression. The differential diagnosis is outlined in Table 2.
TABLE 2 Differential Diagnosis of the Patch and Plaque Stage of Cutaneous T-Cell Lymphoma
| Psoriasis | |
| Atopic dermatitis/eczema | |
| Chronic contact dermatitis | |
| Chronic, nonspecific dermatitis | |
| Drug eruption | |
| Potential precursor lesions | |
| Poikiloderma vasculare atrophicans | |
| Large-plaque parapsoriasis | |
| Small-plaque parapsoriasis | |
| Follicular mucinosis (alopecia mucinosa) | |
| Leukemic variant | |
| Sézary syndrome | |
TREATMENT
The slow evolution of CTCL, wide variability of progression and insidious extracutaneous spread make a specific choice of treatment difficult in many patients. Topical therapies are desirable in patients with limited skin involvement. The alkylating agent mechlorethamine hydrochloride, or nitrogen mustard, may be effective in treating CTCL. Treatment with psoralen plus ultraviolet A (PUVA) may also improve skin lesions in patients with early disease.9
Patients with more extensive disease require systemic therapy to prolong survival. Infection is the usual cause of mortality in patients who die of this disease. The normal T-cell component is eliminated as the disease progresses, and patients become immunosuppressed, not unlike patients with advanced acquired immunodeficiency sydrome.5
Maintaining an index of suspicion for the disease, performing a skin biopsy and vigilant patient follow-up are essential to effectively treat this disorder in its early stages and prevent progression to a life-threatening malignancy.
