Mastocytosis is characterized by an excessive number of apparently normal mast cells in the skin and, occasionally, in other organs. Characteristic skin lesions, called urticaria pigmentosa, are present in most patients, but clinical presentation can vary from a pruritic rash to unexplained collapse and sudden death. These lesions are typically tan to red-brown macules that appear on the trunk and spread symmetrically. Patients with mastocytosis often have a long history of chronic and acute symptoms that were unrecognized as mastocytosis. Skin lesions may or may not accompany systemic mastocytosis. Systemic disease may involve the gastrointestinal tract, the bone marrow or other organs. Even when the disease is considered as a possibility by the physician, the diagnosis can be difficult because of special technical requirements necessary for biopsy and because of the problems with biochemical testing. Drug therapy is initiated to stabilize mast cell membranes, to reduce the severity of the attacks and to block the action of inflammatory mediators. The mainstay of therapy is histamine H1 and H2 blockers and the avoidance of triggering factors.
The diagnosis of mastocytosis is easily missed and often delayed until years after onset, sometimes resulting in a disastrous outcome. Mastocytosis is characterized by an overabundance of normal, active mast cells. The increase in mast cells is usually localized to the skin, but occasionally occurs in other organs as a systemic disorder. Mastocytosis can present at any age, from the newborn period onward. Symptoms of mastocytosis occur when pharmacologic or physical stimuli cause mast cell degranulation and release of histamine, prostaglandins, leukotrienes and other chemical mediators. These episodic attacks can be manifested by flushing, urticaria or, in extreme cases, by life-threatening vascular collapse.
Disease Classification and Epidemiology
A classification scheme for mastocytosis is outlined in Table 1.1 The increase in mastocytes in the skin may be (1) generalized, as in diffuse and erythrodermic mastocytosis; (2) more discrete collections of cells in multiple nests, as in urticaria pigmentosa and telangiectasia macularis eruptiva perstans; or (3) a single collection of cells, as in a solitary mastocytoma. Occurring less frequently than cutaneous forms of mastocytosis are extracutaneous forms, with an increased number of mastocytes in the bone marrow, gastrointestinal tract, liver and spleen, central nervous system, heart or blood.
The prevalence of mastocytosis in the general population is unknown. It has been reported to be present in one of 1,000 to 8,000 new patients evaluated at a dermatology clinic.2 Mastocytosis occurs in all races, and there is no sex predilection. The peak incidence is during infancy and early childhood, with a second peak occurring in middle age. The disease may be benign, with minor transient symptoms and signs that never cause the patient to consult a physician, or it may be life-threatening.3,4
Illustrative Case
Many patients with mastocytosis have a long history of chronic symptoms that are occasionally punctuated by acute exacerbations. In one series of adult patients, the average interval between the onset of symptoms and the correct diagnosis was 10 years.5 The following case illustrates how easily the diagnosis of mastocytosis can be overlooked.
A 53-year-old patient saw her dentist because of a dental abscess and was prescribed penicillin. Before leaving the dentist's office she took two penicillin tablets. She then walked to the parking lot and got into her automobile, which had been sitting in the hot sun. She drove five minutes to her mother's house. The patient reported that while she was driving she felt nauseated and “like I was in a tunnel.” Immediately after she arrived at her mother's house, she lost consciousness. Her mother telephoned 911.
The rescue team found the patient flushed, diaphoretic, wheezing and unresponsive. Her systolic blood pressure was 80 mm Hg. Oxygen, intravenous fluids and epinephrine were administered, and medical antishock trousers (MAST) were applied.
The patient was awake, alert and anxious at the time of admission to the emergency department. She had a palpable blood pressure of 52 mm Hg. She stated that she noticed no shortness of breath, chest pain or a pressure sensation in her chest when she lost consciousness at her mother's house. An electrocardiogram in the emergency department demonstrated diffuse ST-segment changes that were suggestive of ischemia. A dopamine infusion was started, along with methylprednisolone, cimetidine, diphenhydramine and metoclopramide. Two doses of epinephrine were also administered. Her blood pressure increased to 90/50 mm Hg. A repeat electrocardiogram revealed marked improvement. She was admitted to the critical care unit for continued blood pressure support and to exclude a myocardial infarction.
On further questioning, the patient described a similar episode of loss of consciousness four years previously, although that episode was not associated with penicillin. A series of near-syncopal attacks led to a hospital admission, but she was discharged without a definitive diagnosis. She reported having a rash, which had been present for seven to eight years and would become erythematous and pruritic when she was exposed to heat and during emotional stress. She took no medicines and reported no medication allergies, having taken penicillin in the past without incident.
The patient's blood pressure on admission to the critical care unit was 90/55 mm Hg; she was receiving dopamine in a dosage of 5 μg per minute. Her pulse was 94; respiratory rate was 14; and she was afebrile and not in distress.
Cardiac and pulmonary examinations revealed no abnormalities. Hepatosplenomegaly was not found on abdominal examination. Skin examination revealed a macular rash, with tan and salmon-colored lesions most notable on the buttocks, trunk and thighs (Figure 1). The pigmented skin developed a wheal and flare reaction when it was stroked (Darier's sign). Laboratory values were normal except for a potassium level of 2.8 mEq per L.
FIGURE 1.
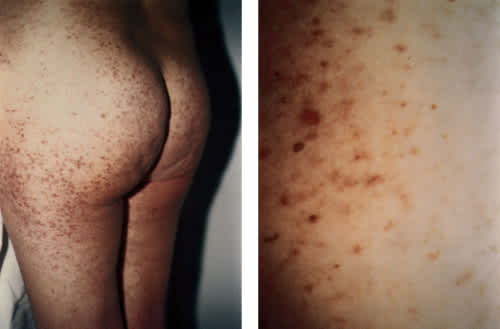
(Left) Cutaneous manifestations of mastocytosis in the patient in the illustrative case. (Right) Close-up view of skin lesions.
By the next morning the patient had been weaned off dopamine, myocardial infarction had been excluded and the ischemic changes on her electrocardiogram had resolved. An echocardiogram and a thallium stress test were normal. A skin biopsy at the site of her rash was first reported as mild inflammation. The pathologist was then informed of the clinical diagnosis. A Giemsa stain confirmed the diagnosis of mastocytosis.
The presumptive diagnosis at the time of hospital discharge was cutaneous mastocytosis. Discharge medications included cimetidine, loratidine and an adrenaline pen. She was instructed to avoid alcohol, penicillin, aspirin and extremes of heat.
Pathophysiology
Mast cells originate from bone marrow progenitor cells and are distributed throughout the connective tissues. They are concentrated in the skin around the peripheral nerves and adjacent to blood and lymphatic vessels.
When activated by IgE or other stimuli, mast cells release preformed mediators of inflammation. These mediators initiate a leukocyte–cytokine cascade that contributes to the acute and delayed hypersensitivity reactions and the various cutaneous and systemic manifestations of mastocytosis (Table 2). The concentration of the mediators varies from site to site, as does the stimulus required for their release at different anatomic sites.6 Many drugs, toxins and physical stimuli can cause mast cell activation (Table 3).2
TABLE 2 Mast Cell Products and Their Clinically Significant Activity
| Mast cell product | Clinically significant activity | |
|---|---|---|
| Preformed secretory granule-associated mediators | ||
| Histamine | Vasodilatation, erythema, edema, pruritus, urticaria, bronchoconstriction, increased gastric acid, intestinal cramping, further degranulation of mast cells, leukocyte activation | |
| Proteoglycans | ||
| Heparin | Osteoporosis, inhibition of localized clotting; rarely, prolonged partial thromboplastin time | |
| Neutral proteases | ||
| Tryptase | Inhibition of coagulation locally, bronchoconstriction, osteoporosis | |
| Chymase | Inhibition of coagulation locally, activation of mast cells, blistering (?) | |
| Cathepsin G and carboxypeptidase | Kinin generation, hepatic fibrosis (?) | |
| Acid hydrolases | Bone lesions, osteoporosis | |
| Lipid mediators | ||
| Leukotrienes | Bronchoconstriction, increased vascular permeability and contractility | |
| Prostaglandin D2 | Pruritus, pain, rhinorrhea, hypotension, flushing, osteoporosis | |
| Platelet-activating factor | Wheal and flare, pain, pruritus | |
| Cytokines | ||
| Tumor necrosis factor | Recruitment of inflammatory cells | |
| Interleukins | Chemoattractant for neutrophils | |
Cutaneous Mastocytosis
Cutaneous mastocytosis constitutes 90 percent of cases of mastocytosis that are not associated with a hematologic disorder.7 Urticaria pigmentosa is the most common variety, occurring in about two thirds of patients with cutaneous mastocytosis.8 It may be apparent at birth, it may appear during the first few months of infancy or it may have an onset at any age thereafter. About one third of patients first develop the characteristic lesions in adulthood.9,10
URTICARIA PIGMENTOSA
The tan to red-brown macules typical of urticaria pigmentosa initially emerge on the trunk and rapidly spread symmetrically and centripetally. The palms, soles, face and scalp are usually spared. The lesions may remain small and freckle-like or may evolve into papules, nodules or plaques. Mucous membranes may also be involved.2 Exposure to physical stimuli such as heat, cold or pressure is typically followed by the onset of localized urticaria (Darier's sign). Local or generalized pruritus is common after exposure to physical stimuli, as is dermatographism, localized erythema and generalized flushing. Vesicles, bullae (rarely with local hemorrhage) and superficial erosions occur only in patients under two years of age.8 Scarring is unusual unless superinfection occurs.
Abdominal pain, nausea, vomiting and diarrhea severe enough to cause malabsorption can occur in patients with a heavy mast cell burden. Headache, hypotension leading to syncope, bleeding diathesis, peptic ulcer disease and psychologic changes such as irritability and difficulty in concentration may also be associated with the disorder.
When urticaria pigmentosa begins in infancy or early childhood, the skin lesions and symptoms eventually abate by adulthood in 50 percent of patients.9 Others show significant improvement, with lesions clearing and symptoms decreasing in frequency and severity. Residual pigmentation can remain after regression of childhood disease. Unlike the childhood disease, adult-onset urticaria pigmentosa usually persists for the patient's lifetime.5
SOLITARY MASTOCYTOMAS
Solitary mastocytomas are more common in infants and children than in adults. They are rare in adults because resolution usually occurs early in life.9 The nodule is usually large (3 to 4 cm), frequently occurs on an extremity and demonstrates Darier's sign. Since the increase in the number of mast cells is small, systemic symptoms are unusual. Second mastocytomas are also uncommon; when they develop, they usually do not occur more than two months after the first lesion.
DIFFUSE ERYTHRODERMIC MASTOCYTOSIS
Diffuse erythrodermic mastocytosis is a rare form of cutaneous mastocytosis that primarily occurs in children under three years of age. The infant's skin may appear normal at birth, but the skin rapidly evolves into a generalized thickening, much like peau d'orange. Bullae and blisters are common, and systemic symptoms can be severe. Spontaneous resolution is the rule, although there is a risk of persistence and systemic involvement extending into adulthood.
TELANGIECTASIA MACULARIS ERUPTIVA PERSTANS
Telangiectasia macularis eruptiva perstans is a rare condition found primarily in adults. Its name describes the nonpruritic lesions of this type of mastocytosis. The lesions in this disorder are smaller than those of urticaria pigmentosa.
Systemic Mastocytosis
Skin lesions may or may not accompany systemic mastocytosis. Systemic involvement can precede, but more frequently follows, the development of urticaria pigmentosa, which is a feature in 85 percent of patients with systemic mastocytosis.5 The prevalence of systemic involvement increases with age, and approaches 15 to 30 percent in adults with skin lesions.10,11
Patients with systemic disease frequently report intermittent, brief spells of urticaria, flushing, pruritus, palpitations, headache, lightheadedness and syncope, rhinorrhea, nausea, vomiting, diarrhea and crampy or burning abdominal pain in association with the ingestion of hot, cold or spicy foods. Gastritis, esophagitis and peptic ulcer disease are common. A period of lethargy usually follows the attacks.
Asymptomatic hepatosplenomegaly with mast cell infiltration is frequently present. Lymphadenopathy can occur. After the skin, the bone marrow is the second most frequently involved organ, and its infiltration by mast cells can lead to bone pain, anemia and pathologic fractures. Other bony lesions include osteosclerosis and osteoporosis.
Diagnosis
Recognition of mastocytosis can be difficult, especially in patients who do not have the characteristic skin lesions and Darier's sign. In addition, many clinicians may be unfamiliar with the disease and thus may fail to consider the diagnosis. Even when mast cell disease is suspected, confirmatory testing can be falsely negative as a result of technical problems.
The three criteria to consider for establishing the diagnosis of mastocytosis are as follows: (1) histopathology of a skin lesion, if available; (2) histologic evidence of systemic involvement with or without an underlying hematologic disorder; and (3) biochemical markers of mast cell activity. The presence of all of these criteria in each patient is not required.
BIOPSY EVIDENCE
When skin lesions are present, a skin biopsy should be obtained to confirm the diagnosis of mastocytosis, since Darier's sign is not pathognomonic. The skin should be anesthetized by injecting a local anesthetic without epinephrine below the biopsy site, not directly into the biopsy site. Direct trauma or epinephrine injection can cause degranulation of the mast cells, making their identification more difficult. Special stains such as toluidine blue, Giemsa or fluorescein isothiocyanate-avidin12 are required because routine histologic staining may not clearly demonstrate the excess number of mast cells. When the specimen is submitted, the pathologist should be informed of the tentative diagnosis.
A liver or bone marrow biopsy in patients with systemic mastocytosis often demonstrates an increase in mast cells. Before a bone marrow sample is obtained, the pathologist must be informed about the possible diagnosis, because a bone marrow sample has special fixation requirements for the diagnosis of mastocytosis.
LABORATORY ABNORMALITIES
Laboratory abnormalities associated with systemic mastocytosis include hypocholesterolemia (10 to 20 percent of patients10), which is probably due to an increased heparin level. Thrombocytopenia, eosinophilia (12 to 25 percent of patients5) and elevation of alkaline phosphatase and liver enzymes may also be present in patients with systemic disease. If there are no characteristic pigmented lesions, a bulla should be biopsied if one is present.
Circulating biochemical mediators are often increased, especially when a large number of mast cells are activated during attacks. Although several biochemical tests of the blood and urine have been used to support the diagnosis of mastocytosis, one of the most sensitive, when it is available, is an elevated plasma tryptase level.4,13 Other indicators of mast cell degranulation are the presence of chronically elevated plasma and urinary histamine levels and its metabolite N-methylhistamine, the presence of chronically elevated urinary metabolites of prostaglandin D214 and a prolonged partial thromboplastin time documented in a blood sample obtained immediately after an attack.15 Serum heparin levels are not useful.2 Testing is not necessary in patients with cutaneous disease only.7
OVERVIEW OF THE DIAGNOSTIC WORK-UP
A suggested diagnostic work-up for adults with suspected mastocytosis is summarized in Table 4. Cutaneous mastocytosis in children is a benign disease. Thus, evaluation beyond a skin biopsy is not necessary. If systemic signs are present or if the disease first appears after age five, the diagnostic work-up in a child is the same as that suggested for an adult.
TABLE 4 Suggested Diagnostic Evaluation of Adults with Suspected Mastocytosis
| For patients with cutaneous mastocytosis, the following studies are recommended: | |
| Skin biopsy | |
| Complete blood count and differential | |
| Bone marrow biopsy | |
| If systemic involvement is suspected, consider obtaining the following studies, depending on the symptoms: | |
| Urine collection for biochemical testing | |
| Bone scan | |
| Gastrointestinal endoscopy | |
| Neuropsychiatric examination | |
| Liver biopsy | |
| Computed tomography of the abdomen | |
When cutaneous mastocytosis begins in adulthood, a bone marrow biopsy is recommended to exclude the presence of an associated hematologic disease, especially in an elderly patient with systemic symptoms, lymphadenopathy or hepatosplenomegaly. A technetium bone scintiscan can establish a baseline to avoid confusion with other bone diseases.2 Bone densitometry should be performed to determine whether osteoporosis is present. Osteoporosis in patients with mastocytosis is believed to be secondary to elevated prostaglandin D2 and heparin levels.
Endoscopy is warranted in patients who have upper gastrointestinal symptoms unresponsive to treatment. Bone marrow biopsy and biochemical tests are required for the diagnosis of systemic mastocytosis in patients without skin involvement. The risk of associated malignant disease in adults with mastocytosis ranges from 2 percent in those with only urticaria pigmentosa9 to 70 percent in elderly symptomatic patients with systemic mastocytosis and no skin lesions.12 The role of periodic surveillance is uncertain, but it may be useful to obtain a complete blood count regularly.
Treatment
Patients with symptomatic mastocytosis should identify and avoid triggering factors. Extremes of temperature, alcohol and certain drugs (Table 3) should be avoided completely or used with caution.
Pharmacologic treatment of mastocytosis involves stabilizing mast cell membranes to decrease the severity of the attacks while blocking the action of inflammatory mediators. Other approaches include surgical removal of isolated mastocytomas, topical corticosteroid therapy and therapy with psoralen plus ultraviolet A (PUVA) to fade multiple pigmented lesions. There is no specific cure for the disease, although improvement can be expected in children.
Histamine H1 antagonists are used to treat the pruritus, flushing, bullae and urticaria. Hydroxyzine (Atarax), doxepin (Sinequan) and chlorpheniramine may help relieve the symptoms. Nonsedating antihistamines may also be used. The dosage is titrated to obtain the most effective regimen. The nonulcer dyspepsia that sometimes occurs with mastocytosis frequently responds to use of H1 blockers.16
Histamine H2 blockers can be used to decrease the gastric hyperacidity that often accompanies concomitant ulcer disease in patients with systemic mastocytosis. Not all mast cells have H1 receptors, and H2 blockers may also decrease cutaneous symptoms.
Orally administered cromolyn sodium (Gastrocrom) can relieve diarrhea and abdominal pain. Although intestinal absorption of cromolyn is limited, this agent has been reported to decrease bone pain and headaches17 while improving cognitive abilities and skin symptoms.8 The dosage is 200 mg four times a day. Several weeks of therapy may be required before benefits are noted. Anticholinergic agents may help alleviate abdominal cramping.
Both H1 and H2 blockers should be used to treat anaphylaxis.18 Patients with mastocytosis should also wear a medical alert bracelet and carry an adrenaline-filled syringe. The emergency treatment of shock in association with mastocytosis is the same as that of anaphylaxsis. Fluids, adrenaline, antihistamines and pressor agents are often required.3
Aspirin has been used to treat the flushing that is associated with elevated prostaglandin levels during attacks. Caution is required, however, because of the potential for aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) to cause degranulation. Treatment with aspirin is reserved for use in patients with vascular collapse that is not prevented by H1 and H2 blocker prophylaxis.19 Treatment is initiated in the hospital, and should begin at a low dosage. The patient should receive antihistamines before intervention with aspirin is undertaken.20
PUVA therapy may provide cosmetic skin changes and temporary relief from symptoms.7,18 High-potency topical or intralesional injection of corticosteroids affords only transient improvement. Topical or intralesional corticosteroid therapy is, however, useful in the treatment of solitary mastocytoma. Systemic steroids may be necessary to control the severe skin disease, malabsorption or ascites of systemic mastocytosis.18,21
