Kawasaki disease is a leading cause of acquired heart disease among children in the United States and other developed countries. Most children who contract this illness are less than two years old, and 80 percent of affected children are younger than five years of age. A generalized vasculitis of unknown etiology, Kawasaki disease can cause coronary artery abnormalities, including coronary aneurysms. From 20 to 25 percent of untreated children develop coronary artery abnormalities, which may resolve or persist. These abnormalities are of particular concern because they can lead to thrombosis, evolve into segmental stenosis or, rarely, rupture. The principal cause of death from Kawasaki disease is myocardial infarction. The cause of the disease remains unknown, but epidemiologic investigations and the clinical presentation suggest a microbial agent. Diagnostic criteria, including fever and other principal features, have been established. In the acute phase of the disease, treatment with acetylsalicylic acid and intravenously administered immunoglobulin is directed at reducing inflammation of the coronary arteries and myocardium. Early recognition and treatment of Kawasaki disease can reduce the development of potentially life-threatening coronary artery abnormalities.
Kawasaki disease or syndrome is a generalized vasculitis of unknown etiology and a leading cause of acquired heart disease among children living in developed countries. First described in 1967 by Kawasaki, this illness was originally referred to as “mucocutaneous lymph node syndrome.”1 Based on findings in 50 Japanese children, Kawasaki described a unique illness that was characterized by fever, rash, conjunctival injection, cervical lymphadenitis, inflammation of the lips and oral cavity, and redness and swelling of the hands and feet. Although initially thought to be a benign childhood illness, the disease was found to be responsible for the death of a number of Japanese children, primarily under two years of age, who had appeared to be improving or to have recovered from the illness.2 Autopsies demonstrated thrombosed occlusion of coronary artery aneurysms and resultant myocardial infarction. It is now known that coronary artery abnormalities develop in approximately 20 to 25 percent of children with untreated Kawasaki disease.3–5
Epidemiology
Kawasaki disease has been reported throughout the world.6 In the United States, the disease has been increasingly recognized since the 1970s, and several regional outbreaks have been reported since 1976.7–10 Kawasaki disease occurs more often in boys than in girls (ratio of about 1.5:1). Approximately 80 percent of affected children are less than five years old. Fewer than 2 percent of children have recurrences.11
The disease occurs year-round, but a greater number of cases are reported in the winter and spring.12–14 Annual incidence rates in the United States and Canada range from about six to 11 cases per 100,000 children less than five years old.10,12,15 Each year in this country, as many as 3,500 children are hospitalized because of Kawasaki disease.15,16 Although the absolute number of U.S. cases is greatest in white children, the incidence rates in North America are highest in children of Asian ethnicity (especially those of Japanese or Korean background).6
In Japan, 12 nationwide epidemiologic incidence surveys on Kawasaki disease have been conducted at two-year intervals beginning in 1970. At the end of the 12th survey in 1992, 116,848 children had been reported to have contracted Kawasaki disease.17 The yearly incidence rate for 1991–1992 was 90 cases per 100,000 children less than five years old.18 About 1 percent of affected children were found to have a family history of sibling Kawasaki disease.18 More than one half of the sibling cases developed within 10 days after the first case.
Etiology
Epidemiologic features and clinical presentation suggest an infectious etiology for Kawasaki disease. To date, however, an etiologic agent has not been documented. A variety of infectious agents have been proposed, including rickettsiae, viruses (primarily Epstein-Barr virus and retroviruses), Streptococcus viridans, staphylococci, Propionibacterium species and parvovirus. Standard laboratory studies have been unsuccessful in identifying a specific agent.19
One study implicated superantigens (i.e., toxins produced by certain staphylococci and streptococci) and proposed a variant of the toxic shock syndrome–associated toxin-1 produced by Staphylococcus aureus as a possible cause of Kawasaki disease.20 To date, however, the findings of this study have not been substantiated.21,22
Even though the initiating event has not yet been identified, the immune system is known to be involved during the acute stage of Kawasaki disease. In response to an unknown triggering process, marked immunoregulatory abnormalities are observed. It is postulated that the various secreted cytokines target vascular endothelial cells, producing cell-surface neoantigens. Antibodies produced against these antigens may then target the vascular endothelium, resulting in a cascade of events leading to vascular damage.
Diagnosis
In the absence of a specific diagnostic test, Kawasaki disease is a clinical diagnosis based on the characteristic history and physical findings. Clinical criteria were developed by the Japan Kawasaki Disease Research Committee23 and subsequently by the American Heart Association (AHA).24 These criteria are classified as principal clinical findings and other significant clinical and laboratory findings.
The principal clinical findings in Kawasaki disease are listed in Table 1.25 Fever and at least four of the five principal clinical findings generally should be present to establish the diagnosis. However, “atypical” or “incomplete” cases of Kawasaki disease, in which patients have fewer than four of the five principal features, have been increasingly reported. Classic diagnostic criteria are more often lacking in infants, especially in those less than six months old. Patients with fever and three of the principal clinical features can be diagnosed with Kawasaki disease when coronary artery disease is detected by two-dimensional echocardiography or coronary angiography.
TABLE 1 Diagnostic Criteria for Kawasaki Disease: Principal Clinical Findings
| Fever persisting at least five days*: the fever is generally high and spiking (often to 40°C [104°F] or higher) and persists in untreated patients for one to two weeks or longer. | |
| Presence of at least four of the following five principal features†: | |
| Changes in extremities: these changes are distinctive and acutely include redness, swelling and, sometimes, induration of the hands and feet. One to three weeks after the onset of fever, desquamation of the fingers and toes occurs. Approximately one to two months after the onset of fever, Beau's lines (white lines across the fingernails) may appear. | |
| Polymorphic exanthem: the skin eruption involves the trunk and extremities and may have several forms, including urticarial exanthem, a morbilliform maculopapular eruption (occasionally with target lesions) or a diffuse scarlatiniform rash. Bullae and vesicles are not seen. The rash usually appears within five days after the onset of fever. | |
| Bilateral conjunctival injection: the bulbar conjunctivae, rather than the palpebral or tarsal conjunctivae, are involved. Typically, the limbic region is spared. The conjunctival injection is not associated with an exudate and is usually painless. | |
| Changes in the lips and oral cavity: these changes include strawberry tongue, redness and cracking of the lips, and erythema of the oropharyngeal mucosa. Ulcerative lesions are not seen. | |
| Cervical lymphadenopathy (at least one lymph node with a diameter of 1.5 cm or greater): the lymphadenopathy is usually unilateral, with firm and slightly tender nodes. | |
| Exclusion of other diseases with similar findings. | |
*—Many experts believe that when the classic features are present, Kawasaki disease can be diagnosed by experienced observers before day 5 of fever.
†—The first four of the five principal clinical findings are present in about 90 percent of patients with Kawasaki disease. Cervical lymphadenopathy is observed in 50 to 75 percent of patients.
Adapted from Dajani AS, Taubert KA, Gerber MA, Shulman ST, Ferrieri P, Freed M, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation 1993;87:1776–80.
A high index of suspicion is required to diagnose Kawasaki disease in patients who have a number of the classic features, but not enough of them to fulfill the diagnostic criteria. Some of these patients have prolonged unexplained fever or cervical lymphadenopathy that does not respond to antibiotic therapy, or they may have fever with two to three of the classic criteria for Kawasaki disease. It has become clear that patients with atypical or incomplete Kawasaki disease are at substantial risk for the development of coronary abnormalities26 and therefore would benefit from treatment if they could be identified.
Because the principal clinical findings that fulfill the diagnostic criteria for Kawasaki disease are not specific, other diseases and disorders with similar clinical findings should be ruled out (Table 2).25 Careful consideration of measles is particularly important because appropriate control measures cannot be taken promptly if this illness is misdiagnosed as Kawasaki disease.
TABLE 2 Selected Differential Diagnosis of Kawasaki Disease
| Measles |
| Scarlet fever |
| Drug reactions |
| Stevens-Johnson syndrome |
| Other febrile viral exanthems |
| Toxic shock syndrome |
| Rocky Mountain spotted fever |
| Staphylococcal scalded skin syndrome |
| Juvenile rheumatoid arthritis |
| Leptospirosis |
| Mercury poisoning |
Reprinted with permission from Dajani AS, Taubert KA, Gerber MA, Shulman ST, Ferrieri P, Freed M, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation 1993;87:1776–80. Copyright American Heart Association.
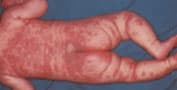
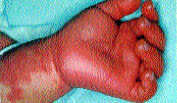
The course of Kawasaki disease can be divided into three clinical phases: acute, subacute and convalescent. The acute febrile phase usually lasts seven to 14 days. This phase, which ends with the resolution of fever, is characterized by conjunctival injection, mouth and lip changes, swelling and erythema of the hands and feet, rash and cervical lymphadenopathy (Figures 1, 2 and 3).27 The subacute phase covers the period from the end of the fever to about day 25. During this phase, patients may have desquamation of the fingers and toes (Figure 4),27 arthritis and arthralgia, and thrombocytosis. The convalescent phase begins when clinical signs disappear and continues until the erythrocyte sedimentation rate becomes normal, usually six to eight weeks after the onset of illness.
FIGURE 1.
Rash on day 4 of illness in a seven-month-old boy with Kawasaki disease.
FIGURE 2.

Conjunctival injection, lip edema and erythema on day 6 of illness in a two-year-old boy with Kawasaki disease.
FIGURE 3.
Edema and erythema on day 6 of illness in a one and one-half–year-old girl with Kawasaki disease.
FIGURE 4.
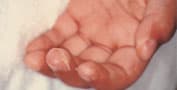
Periungual desquamation on day 12 of illness in a three-year-old child with Kawasaki disease.
Figures 1 through 4 used with permission from Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the AHA's Council on Cardiovascular Disease in the Young. Diagnostic guidelines for Kawasaki disease. Dallas: American Heart Association, 1993. Copyright American Heart Association.
Clinical and Laboratory Findings
Findings not included in the major diagnostic criteria are often present in patients with Kawasaki disease. These are subdivided into cardiac, noncardiac and laboratory findings.
CARDIAC FINDINGS
Cardiovascular manifestations can be prominent in the acute phase of the illness and are the leading cause of morbidity and mortality (Table 3).28 Pancarditis may occur, and the coronary arteries may be affected.3 Pericardial effusion, which usually resolves spontaneously without specific therapy, is detected by echocardiography in approximately 30 percent of patients.3 Clinically recognizable myocarditis, occasionally with congestive heart failure, is commonly manifested by tachycardia out of proportion to the degree of fever, a gallop rhythm or arrhythmias. Electrocardiographic changes are nonspecific and are observed in one third of patients. These changes include decreased R-wave voltage, ST-segment depression with T-wave flattening or inversion and a prolonged PR or QT interval (or both).3
Coronary artery abnormalities develop in 20 to 25 percent of children with untreated Kawasaki disease.3–5 Abnormalities may include diffuse ectasia or coronary aneurysms (Figures 5 and 6).27 Patients who have giant aneurysms (maximum internal diameter of at least 8 mm) have the worst prognosis and are at greatest risk of developing coronary thrombosis, stenosis or myocardial infarction. Unlike smaller aneurysms, these giant aneurysms generally do not resolve. Children with coronary artery abnormalities, particularly giant coronary aneurysms, are more likely to have aneurysms of other medium-sized muscular arteries, such as renal, brachial or femoral arteries. Aneurysms usually become apparent one to three weeks after the onset of fever; their appearance more than five weeks after the onset of illness is uncommon. Risk factors for coronary aneurysms in patients with Kawasaki disease are listed in Table 4.25
FIGURE 5.
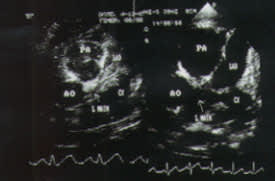
Two-dimensional echocardiograms. (Left) Very small normal vessels (arrows). (Right) Abnormally enlarged vessels (arrows). (AO = aorta; PA = pulmonary artery; LAD = left anterior descending coronary artery; CX = circumflex coronary artery; L Main = left main coronary artery)
FIGURE 6.
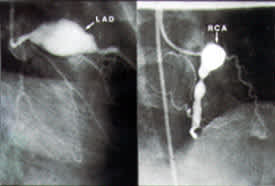
Coronary angiograms in a six-year-old boy with Kawasaki disease. (Left) Hugely dilated left descending (LAD) artery with obstruction. (Right) Very dilated right coronary artery (RCA) with an area of severe narrowing.
Figures 5 and 6 used with permission from Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the AHA's Council on Cardiovascular Disease in the Young. Diagnostic guidelines for Kawasaki disease. Dallas: American Heart Association, 1993. Copyright American Heart Association.
TABLE 4 Risk Factors for Coronary Aneurysms in Kawasaki Disease
| Male gender |
| Age less than one year |
| Other signs or symptoms of pericardial, myocardial or endocardial involvement, including arrhythmias |
| Prolonged period of inflammation, including fever for more than 10 days |
| Recurrence of fever after an afebrile period of at at least 24 hours |
Information from Dajani AS, Taubert KA, Gerber MA, Shulman ST, Ferrieri P, Freed M, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation 1993;87:1776–80.
Echocardiograms and angiograms obtained in long-term follow-up studies indicate that coronary aneurysms resolve within five to 18 months in approximately 50 percent of patients.4 In these studies, one half of the patients with persistent abnormalities showed decreased size of aneurysms, one third had resolution of aneurysms but developed coronary obstruction or stenosis, and the remainder had fine irregularities of the coronary walls without stenosis. Myocardial ischemia may occur in the presence of stenosis with obstruction.
If not treated within the first 10 days of illness, patients with Kawasaki disease who are younger than one year (especially those younger than eight months) have a substantially increased risk of developing coronary artery aneurysms.29 Compared with older children, young infants tend to have subtle manifestations of Kawasaki disease. Consequently, the presence of this illness is often more difficult to suspect and confirm in very young patients.
NONCARDIAC FINDINGS
Several noncardiac clinical findings may be observed in patients with Kawasaki disease (Table 5).28 For example, infants and young children with this disease are often more irritable than children with other febrile illnesses. Arthritis and arthralgia (more common in older girls) can occur in the first two to three weeks of the illness and typically involve the knees, ankles or hips. About one fourth of patients who undergo lumbar puncture in the acute phase of Kawasaki disease have findings consistent with aseptic meningitis. Other common features include diarrhea, vomiting, abdominal pain and pneumonitis.
Acute acalculous distention of the gallbladder (hydrops) may occur during the first two weeks of illness. Recently, elevated serum bile acid levels were reported in patients with acute Kawasaki disease.30 Less common findings include erythema and induration at the site of recent bacille Calmette-Guérin vaccination. Auditory abnormalities, testicular swelling and peripheral gangrene have also been reported in patients with the disease.
LABORATORY FINDINGS
Laboratory findings and their frequency in Kawasaki disease are listed in Table 6.28 Although these findings are nonspecific for Kawasaki disease, they may assist in establishing the diagnosis. During the first seven to 14 days of the disease, patients often have increased acute-phase reactants; these values then return to normal within six to eight weeks. Neutrophilia, mild anemia, hypoalbuminemia and elevated serum immunoglobulin E levels may also be observed. Thrombocytosis is frequently present after the first week of illness and may be marked. Proteinuria and sterile pyuria of urethral origin are also common.
Treatment
In the absence of a known etiologic agent, initial therapy for Kawasaki disease is directed at reducing fever and other inflammatory features to help prevent the development of coronary artery abnormalities and subsequent myocardial ischemic injury. Long-term therapy is targeted at avoiding coronary thrombosis by preventing platelet aggregation.
Current recommendations for initial therapy (preferably given within the first 10 days of the onset of illness) include the intravenous administration of immunoglobulin to help prevent coronary artery abnormalities and the oral administration of acetylsalicylic acid (aspirin) in high dosages to hasten resolution of the acute manifestations of Kawasaki disease, especially fever (Table 7).25 The AHA recommends that aspirin be given in a dosage of 80 to 100 mg per kg per day during the acute phase of the disease. This dosage should be continued until the patient has been afebrile for at least several days. The dosage is then reduced to 3 to 5 mg of aspirin per kg per day and continued for its antithrombotic effect. Aspirin therapy can be discontinued after six to eight weeks if echocardiograms show no evidence of coronary artery abnormalities. If coronary artery abnormalities are detected, low-dose aspirin therapy should be continued indefinitely.31 To reduce the very small risk of Reye's syndrome, aspirin therapy should probably be interrupted for a week or so if the patient develops varicella or influenza.
TABLE 7 Recommended Therapy for Kawasaki Disease
| Acute phase | |
| Intravenously administered immunoglobulin, 2 g per kg, as a single infusion over 10 to 12 hours | |
| Plus | |
| Acetylsalicylic acid (aspirin), 80 to 100 mg per kg per day orally in four equally divided doses until the patient has been afebrile for several days* | |
| Subacute and convalescent phases | |
| Aspirin, 3 to 5 mg per kg orally once daily for as long as six to eight weeks† | |
*—Some clinicians recommend the use of high-dose aspirin therapy until day 14 of illness.
†—Aspirin can be discontinued after six to eight weeks if all echocardiograms show no evidence of coronary artery abnormalities. If coronary artery abnormalities are detected, low-dose aspirin therapy should be continued indefinitely.
Adapted from Dajani AS, Taubert KA, Gerber MA, Shulman ST, Ferrieri P, Freed M, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation 1993;87:1776–80.
The exact mechanism of action of intravenously administered immunoglobulin is unknown, but its beneficial effect in preventing coronary artery abnormalities was first reported in 1984.32 The combination of intravenously administered immunoglobulin and oral aspirin has been shown to be superior to aspirin alone in achieving defervescence, reducing inflammation and preventing the development of coronary artery aneurysms,5,32–34 especially giant aneurysms.35 The AHA recommends the use of a single immunoglobulin dose of 2 g per kg; alternatively, a four-day infusion of 400 mg per kg per day may be given.25 One U.S. trial showed that patients who received the single-dose immunoglobulin regimen had faster resolution of symptoms and significantly fewer coronary artery abnormalities two weeks after the start of the treatment than patients who received the four-day regimen.33
Some patients are diagnosed with Kawasaki disease later than 10 days after the onset of illness and already have coronary artery ectasia or aneurysms. Current data are lacking on the long-term benefits of aspirin and immunoglobulin therapy later than day 10 of illness.
Some patients have persistent fever 24 to 48 hours after the immunoglobulin infusion is completed. Other patients may have an initial defervescence for at least 24 hours but become febrile again. In both circumstances, a repeat dose of immunoglobulin (2 g per kg) is generally given, and the patient should be referred to a center experienced in the diagnosis and treatment of Kawasaki disease.
Except in unusual circumstances, corticosteroids appear to be contraindicated in the treatment of Kawasaki disease. Coronary abnormalities have developed significantly more often in patients treated with steroids alone during the acute phase of Kawasaki disease than in patients treated with 30 mg per kg per day of aspirin alone.36
Treatment of Complications
Experience with percutaneous transluminal coronary angioplasty is very limited in patients who have Kawasaki disease. Intracoronary or intravenous thrombolytic therapy has been used with varying efficacy in selected patients. In some patients, coronary artery obstruction may be severe enough to warrant surgical revascularization.37 Worldwide, at least 13 patients with Kawasaki disease have undergone cardiac transplantation because of severe ischemic disease.38
Myocardial infarction is the principal cause of death in patients with Kawasaki disease. Although infarction may occur during the acute phase of the disease, it more commonly occurs a year or even years later, especially in patients with giant aneurysms. Symptoms of myocardial infarction include inconsolable crying, vomiting, dyspnea, cardiovascular collapse and shock. Most documented cases of infarction have occurred during sleep or at rest.39 In patients with Kawasaki disease, the reported mortality rates for a first myocardial infarction range from 14 to 22 percent.39,40 The mortality rate for a second infarction is 16 percent.40
