Systemic vasculitis occurs in a heterogeneous group of primary disorders or can be a manifestation of infection, an adverse drug reaction, malignancy or a connective tissue disease. A vasculitic process should be suspected in patients with unexplained ischemia or multiple organ involvement, especially when such features as polymyalgia rheumatica, inflammatory arthritis, palpable purpura, glomerulonephritis or multiple mononeuropathy are also present. The clinical features of systemic vasculitis depend on the organs involved and, in turn, organ involvement is largely influenced by the size of the affected blood vessels. The diagnostic work-up should be tailored to the clinical situation and geared toward a tissue or angiographic diagnosis, bearing in mind that the findings from these studies are not always pathognomonic. Emphasis should also be placed on exclusion of a secondary process. The diagnosis of the specific type of vasculitis may be made on the basis of the clinical features and the histopathologic or angiographic findings. Initial therapy for most types of systemic vasculitis consists of high-dose corticosteroids, with the addition of immunosuppressive therapy in certain patients.
Vasculitis is a nonspecific term that encompasses a large and heterogeneous group of disorders that are characterized by inflammation of blood vessels. The term “systemic necrotizing vasculitis” describes a systemic process in which blood vessel architecture has been destroyed by inflammatory cells. Confusion over the diagnosis of systemic vasculitis arises from the protean and oftentimes nonspecific clinical manifestations, as well as the inconsistent nomenclature used in the literature.
Understanding Vasculitis
As described by Mandell and Hoffman,1 vasculitis-induced injury to blood vessels may lead to increased vascular permeability, vessel weakening that causes aneurysm formation or hemorrhage, and intimal proliferation and thrombosis that result in obstruction and local ischemia. Because systemic vasculitis can affect vessels of all sizes and distributions, it has a wide spectrum of clinical features. Knowing the size of the vessels affected in a particular patient is important, since vessel size carries implications for the diagnosis, treatment and prognosis of the disease (Table 1).
TABLE 1 Clinical Features and Primary Treatment of the Major Systemic Vasculitis Syndromes
| Systemic vasculitis syndrome | Common presenting features | Primary treatment |
|---|---|---|
| Vasculitis of small vessels | ||
| Hypersensitivity vasculitis | Palpable purpura | Often self-limited if offending agent is removed. If isolated to skin, may not require therapy. In more severe cases, moderate- to high-dose corticosteroid therapy may be needed. |
| Henoch-Schönlein purpura | Palpable purpura, arthritis, glomerulonephritis, intestinal ischemia | Often self-limited and requires no treatment. Steroid therapy for some cases of gastrointestinal or renal involvement. |
| Cryoglobulinemia | Arthritis, Raynaud's phenomenon, glomerulonephritis, palpable purpura | Corticosteroids; plasmapheresis for severe involvement. Antiviral therapy required if associated with hepatitis C. |
| Vasculitis of small and medium-sized vessels | ||
| Polyarteritis nodosa | Peripheral neuropathy, mononeuritis multiplex, intestinal ischemia, renal ischemia, testicular pain, livedo reticularis | High-dose corticosteroids, often with cytotoxic agents (e.g., cyclophosphamide [Cytoxan]) |
| Microscopic polyangiitis | Pulmonary hemorrhage, glomerulonephritis | High-dose corticosteroids, often with cytotoxic agents (e.g., cyclophosphamide) |
| Churg-Strauss vasculitis | Allergic rhinitis, asthma, eosinophilia, pulmonary infiltrates, coronary arteritis, intestinal ischemia | High-dose corticosteroids, often with cytotoxic agents (e.g., cyclophosphamide) |
| Wegener's granulomatosis | Recurrent epistaxis or sinusitis, pulmonary infiltrates and/or nodules, glomerulonephritis, ocular involvement | High-dose corticosteroids and cyclophosphamide. Corticosteroids and methotrexate may be used for less severe involvement. |
| Kawasaki disease | Fever, conjunctivitis, lymphadenopathy, desquamating rash, mucositis, arthritis, coronary artery aneurysms | High-dose aspirin and intravenous immune globulin |
| Vasculitis of large vessels | ||
| Giant cell, or temporal, arteritis | Headache, polymyalgia rheumatica, jaw or tongue claudication, scalp tenderness, fever, vision disturbances | High-dose corticosteroids |
| Takayasu's arteritis | Extremity claudication, athralgias, constitutional symptoms, renal ischemia | High-dose corticosteroids |
It is critical to distinguish vasculitis occurring as a primary autoimmune disorder from vasculitis secondary to infection, drugs, malignancy or connective tissue disease such as systemic lupus erythematosus (SLE) or rheumatoid arthritis.2 Immunosuppressive therapy, which is sometimes used in the treatment of certain types of vasculitis, can have catastrophic consequences in the face of a systemic infection, as can delayed or inappropriate treatment of primary systemic vasculitis. Consequently, much of the diagnostic work-up in a patient with suspected vasculitis is directed at excluding secondary causes or conditions that can mimic vasculitis.
Classification of Vasculitis
Classification of vasculitis has received much attention over the past several decades, but no universally accepted classification system has emerged. Vasculitis may be classified by the size and type of vessel involvement, by the histopathologic features (leukocytoclastic, granulomatous vasculitis, etc.) or by the pattern of clinical features.3
Identifying the type of vasculitis is important, since certain types may be self-limited, whereas others may require corticosteroid therapy, with or without a cytotoxic agent, or other modalities such as plasmapheresis. Initially in the work-up, however, determining the extent of visceral organ involvement is more important than identifying the type of vasculitis, so that organs at risk of damage are not jeopardized if treatment is delayed or inadequate.
The clinical features of primary vasculitis syndromes often overlap, and many patients do not fit neatly into a well-defined type of vasculitis. These patients may be described as having “undifferentiated systemic vasculitis.” Such cases require close follow-up, looking for signs of involvement in other organs or signs that may lead to a specific diagnosis.
When to Suspect Systemic Vasculitis
Table 2 outlines the clinical features that are common in the vasculitides, although some may also occur in chronic malignant processes and infectious diseases.4 In particular, two types of presentation should alert the clinician to the possibility of systemic vasculitis: (1) unexplained ischemia, such as claudication, limb ischemia, angina, transient ischemic attack, stroke, mesenteric ischemia and cutaneous ischemia, particularly in a young patient or a patient without risk factors for atherosclerosis and (2) multiple organ dysfunction in a systemically ill patient, especially in the presence of other suggestive clinical features.5
TABLE 2 Clinical Features That Raise Suspicion of Vasculitis
| General clinical feature | Signs or presenting disorder | Type of vasculitis |
|---|---|---|
| Constitutional symptoms | Fever, fatigue, malaise, anorexia, weight loss | Any type of vasculitis |
| Polymyalgia rheumatica | Proximal muscle pain with morning stiffness | Giant cell arteritis; less commonly, other vasculitides |
| Nondestructive oligoarthritis | Joint swelling, warmth, painful range of motion | Polyarteritis, Wegener's granulomatosis, Churg-Strauss vasculitis |
| Skin lesions | Livedo reticularis, necrotic lesions, ulcers, nodules, digital tip infarcts | Polyarteritis, Churg-Strauss vasculitis, Wegener's granulomatosis, hypersensitivity vasculitis |
| Palpable purpura | Any type of vasculitis except giant cell arteritis and Takayasu's arteritis | |
| Multiple mononeuropathy (mononeuritis multiplex) | Injury to two or more separate peripheral nerves (e.g., patient presents with both right foot drop and left wrist drop) | Polyarteritis, Churg-Strauss vasculitis, Wegener's granulomatosis, cryoglobulinemia |
| Renal involvement | Ischemic renal failure related to arteritis | Polyarteritis, Takayasu's arteritis; less commonly, Churg-Strauss vasculitis and Wegener's granulomatosis |
| Glomerulonephritis | Microscopic polyangiitis, Wegener's granulomatosis, cryoglobulinemia, Churg-Strauss vasculitis, Henoch-Schönlein purpura |
General Approach to Diagnosis
When systemic vasculitis is suspected, the diagnostic work-up can be approached in a stepwise fashion:
- Attempt to exclude other processes, particularly infection, thrombosis and neoplasia, that can cause secondary vasculitis or can have features that mimic vasculitis,5,6 (Table 3). This must be done quickly to provide appropriate therapy for a potentially life-threatening condition. In addition, anti-inflammatory and immunosuppressive therapy of the systemic vasculitides is potentially toxic and may mask certain disorders, such as infection or a perforated viscus.
- Consider the patient's age, sex and ethnic origin, since certain vasculitis syndromes occur more commonly in specific populations7 (Table 4).
- Determine which organs are involved and estimate the size of vessel involvement. The type and extent of organ involvement in vasculitis can be helpful in determining the specific type of vasculitis and the degree of urgency in initiating treatment. The clinical features in a given patient can be used to discern the size of the vessels affected by vasculitis (Table 5). It should be recognized that the terms “small,” “medium” and “large” to describe vessel size have different meanings for a pathologist, an angiographer and a clinician.
- Attempt to distinguish the specific type of vasculitis on the basis of the above information and the pattern of the clinical features.
TABLE 3 Conditions That Can Mimic Primary Systemic Vasculitis
| Embolic disease |
| Endocarditis |
| Atrial myxoma |
| Cholesterol embolization |
| Vessel stenosis or “spasm” |
| Atherosclerosis |
| Fibromuscular dysplasia |
| Drug-induced vasospasm (e.g., ergots, cocaine, phenylpropanolamine) |
| Intravascular lymphoma |
| Vessel thrombosis |
| Disseminated intravascular coagulopathy |
| Thrombotic thrombocytopenic purpura |
| Antiphospholipid antibody syndome |
| Heparin- or warfarin-induced thrombosis |
| Systemic infection |
| Malignancy |
| Other connective tissue disorders |
TABLE 4 Demographic Associations of the Vasculitides
| Age group | Male-to- female ratio | Ethnic origin | Type of vasculitis |
|---|---|---|---|
| Child | M = F | Any | Henoch-Schönlein purpura |
| M > F | Asian > white > others | Kawasaki disease | |
| Young adult | M = F | Middle Eastern > others | Behçet's disease |
| F > M | Asian >> others | Takayasu's arteritis | |
| Middle age | M > F | Any | Wegener's granulomatosis, polyarteritis, Churg-Strauss vasculitis |
| Elderly | F > M | Caucasian >> others | Giant cell arteritis |
TABLE 5 Clues for Identifying the Type of Vessel Involvement in Vasculitis
| Clinical feature | Most likely affected vessel | Most commonly associated systemic vasculitis |
|---|---|---|
| Cutaneous | ||
| Palpable purpura | Postcapillary venules | Any type of vasculitis except giant cell arteritis and Takayasu's arteritis |
| Skin ulcers | Arterioles to small arteries | Polyarteritis, Churg-Strauss vasculitis, Wegener's granulomatosis, hypersensitivity vasculitis |
| Gangrene in an extremity | Small to medium-sized arteries | Polyarteritis, Churg-Strauss vasculitis, Wegener's granulomatosis |
| Gastrointestinal tract | ||
| Abdominal pain or mesenteric ischemia | Small to medium-sized arteries | Henoch-Schönlein purpura, polyarteritis, Churg-Strauss vasculitis |
| Gastrointestinal bleeding | Capillaries to medium-sized arteries | Henoch-Schönlein purpura, polyarteritis, Churg-Strauss vasculitis |
| Renal | ||
| Glomerulonephritis | Capillaries | Microscopic polyangiitis, Wegener's granulomatosis, cryoglobulinemia, Churg-Strauss vasculitis, Henoch-Schönlein purpura |
| Ischemic renal failure | Small to medium-sized arteries | Polyarteritis, Takayasu's arteritis; less commonly, Churg-Strauss vasculitis and Wegener's granulomatosis |
| Pulmonary | ||
| Pulmonary hemorrhage | Capillaries; less commonly small to medium-sized arteries | Microscopic polyangiitis, Wegener's granulomatosis |
| Pulmonary infiltrates or cavities | Small to medium-sized arteries | Wegener's granulomatosis, Churg-Strauss vasculitis, microscopic polyangiitis |
| Neurologic | ||
| Peripheral neuropathy | Small arteries | Polyarteritis, Churg-Strauss vasculitis, Wegener's granulomatosis, cryoglobulinemia |
| Stroke | Small, medium-sized or large arteries | Giant cell arteritis, SLE-associated vasculitis |
SLE = systemic lupus erythematosus.
LABORATORY TESTING
Although occasionally helpful in classifying vasculitis, laboratory testing is most important in determining organ involvement and excluding the presence of other diseases (Table 6). Important routine tests include complete blood cell count, urinalysis, blood urea nitrogen, creatinine and liver enzyme levels. Leukocytosis, anemia of chronic disease, a high erythrocyte sedimentation rate (ESR) and an elevated C-reactive protein level are commonly found in most types of vasculitis. Although ESR is almost always elevated in vasculitis, a normal ESR does not rule out systemic vasculitis.1 If the patient has renal failure or proteinuria and hematuria on urinalysis, a fresh-spun urine sample should be evaluated for red blood cell casts or dysmorphic red cells that would suggest the presence of a glomerulonephritis.
TABLE 6 Laboratory Tests to Consider in the Evaluation of Systemic Vasculitis
| Laboratory test | Purpose or interpretation |
|---|---|
| Routine tests (including complete blood cell count, liver enzymes, creatinine, urinalysis) | Evaluate for hematologic, renal and other organ involvement |
| Blood cultures | Rule out infection |
| Erythrocyte sedimentation rate | High value suggests inflammatory disease |
| C-reactive protein | High value suggests inflammatory disease |
| Rheumatoid factor | Very high titers in rheumatoid arthritis, Sjögren's syndrome and cryoglobulinemia-associated vasculitis |
| Antinuclear antibody | Screen for SLE and Sjögren's syndrome |
| Complements (C3, C4, CH50) | Low complement levels suggest consumption by immune complexes, which are commonly found in SLE and cryoglobulinemia |
| Cryoglobulins | Must be present to diagnose mixed essential cryoglobulinemia but can be found in any primary or secondary vasculitis |
| ANCA | Cytoplasmic ANCA pattern specific for Wegener's granulomatosis; perinuclear ANCA pattern may occur in other vasculitides |
| Creatine phosphokinase | Elevation suggests myositis, which can occur in many vasculitis syndromes |
| RPR/VDRL | Rule out syphilis |
| Serum protein electrophoresis | Evaluate for plasma cell dyscrasias |
| Hepatitis B and C serology | Rule out hepatitis B or hepatitis C infection |
| HIV | Rule out HIV infection |
| Anti-glomerular basement membrane | Rule out Goodpasture's syndrome, which can mimic vasculitis and cause pulmonary hemorrhage and glomerulonephritis |
SLE = systemic lupus erythematosus; ANCA = anti-neutrophil cytoplasmic antibodies; RPR = rapid plasmin reagin; HIV = human immunodeficiency virus.
Chest radiographs may reveal asymptomatic pulmonary involvement. If findings suggestive of peripheral neuropathy or muscle inflammation are noted on the history and physical examination, nerve conduction velocity and electromyographic testing can help confirm these suspicions and serve as a guide for biopsy.
Although rheumatoid factor can be present in any inflammatory disease, it is more often found in rheumatoid arthritis–associated vasculitis, Sjögren's syndrome and mixed cryoglobulinemia. Hepatitis B, hepatitis C and human immunodeficiency virus (HIV) serologies should be obtained, since these infections are sometimes associated with vasculitis.8–10 While complement levels (C3, C4 and CH50) are often normal or high in other types of vasculitis, they tend to be low in SLE-associated vasculitis, cryoglobulinemia and hepatitis B– and C–associated vasculitis, reflecting consumption by circulating immune complexes.
Cryoglobulins can be present in malignancy (plasma cell dyscrasias, lymphoma and leukemia), chronic infections (hepatitis B, hepatitis C and endocarditis), inflammatory rheumatic disease (Sjögren's syndrome, SLE and rheumatoid arthritis) or in the syndrome of mixed essential cryoglobulinemia. Mixed essential cryoglobulinemia is now known to be strongly associated with hepatitis C.9 Cryoglobulins are antibodies that reversibly precipitate in cold. They should be collected in a warmed tube and kept warm on transport to the laboratory.
There are two staining patterns for antineutrophil cytoplasmic antibodies (ANCA). Cytoplasmic ANCA (C-ANCA) most often represents antibody to proteinase 3 and is present in up to 90 percent of patients with active, diffuse Wegener's granulomatosis and quite specific in the appropriate clinical setting.11 Perinuclear ANCA (P-ANCA), which is often caused by antimyeloperoxidase antibody, is less specific but is often present when vasculitis affects small to medium-sized arteries.
ARTERIOGRAPHY OR BIOPSY?
Confirmation of a clinical suspicion of vasculitis usually requires arteriography, biopsy, or both. Evaluation should be directed toward establishing a tissue diagnosis, if possible. In general, because “blind” biopsy of asymptomatic sites or organs generally has a low yield,2 it is best to “go where the money is.” For example, if a patient is over age 50 and presents with a new, unexplained headache and elevated ESR, with or without a tender or abnormal temporal artery, a temporal artery biopsy would be indicated. Similarly, in a patient who presents with a multisystem illness and testicular pain and swelling, a testicular biopsy should be considered. Sural nerve biopsy may be indicated in a patient with numbness and tingling in a lower extremity. If the urine sediment is abnormal, a renal biopsy might be obtained.
If a biopsy is impractical, an angiogram may be diagnostic. An angiogram should be obtained in cases of suspected large-vessel vasculitis, such as of the branches of the aorta, which may be manifested as extremity ischemia or claudication. A patient with abdominal pain may have mesenteric or renal vasculitis. If visceral involvement is suspected (and in the absence of a surgical abdomen), a visceral angiogram to include the celiac, mesenteric, renal and, perhaps, hepatic arteries should be performed (Figure 1).
FIGURE 1.
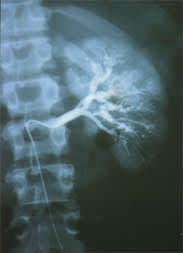
Renal arteriogram in an 18-year-old patient with polyarteritis nodosa. Note areas of vessel dilatation and stenosis, and aneurysms typical of a medium-sized artery vasculitis. Similar findings were also seen in the mesenteric vasculature.
PITFALLS IN DIAGNOSIS
Although biopsy or angiography can be diagnostic for vasculitis, there are a number of potential pitfalls to remember:
- The angiographic appearance of diseases such as fibromuscular dysplasia, atherosclerosis and drug-induced vasospasm can be similar to that of vasculitis and should therefore be interpreted with caution.6
- Small-vessel involvement in the skin or visceral organs can also be the result of immune complex deposition in association with infection, drugs, neoplasia or inflammatory disorders, which are not primary vasculitides.
- A skin biopsy showing leukocytoclastic vasculitis (which occurs in small dermal vessels) does not prove true systemic or visceral involvement nor does it prove or disprove involvement of larger vessels.
- None of the pathologic processes that occur in association with vasculitis are diagnostic of a specific vasculitic syndrome. Rather, the findings must be interpreted with the clinical picture in mind.12
- Vasculitic lesions tend to be focal and segmental, so thorough sectioning of a generously sized specimen may be needed if the initial histopathologic findings are nondiagnostic.
- Finally, a renal biopsy only rarely reveals vasculitis in the renal vessels themselves but may show focal segmental necrotizing glomerulonephritis, a finding that is suggestive of systemic necrotizing vasculitis in the appropriate clinical setting. In addition, renal biopsy findings usually cannot differentiate the type of vasculitis.
A Quick Overview of Selected Vasculitis Syndromes
VASCULITIS OF SMALL VESSELS
Hypersensitivity vasculitis primarily affects postcapillary venules and arterioles of the skin. This disorder usually presents as palpable purpura, although lesions may occasionally be urticarial or ulcerative (Figure 2). Skin biopsy usually shows leukocytoclastic angiitis. Although hypersensitivity vasculitis is occasionally idiopathic, there are multiple known etiologies, including medications, infections, malignancy and primary connective tissue diseases. If hypersensitivity vasculitis is suspected, diagnostic evaluation should focus on identifying an underlying cause and on looking for other organ involvement and large-vessel involvement, which may require more aggressive treatment.
FIGURE 2.
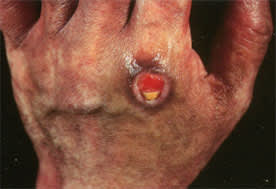
Large vasculitic cutaneous ulcer in a 51-year-old patient with dermatomyositis-associated vasculitis. The size of the lesion and its location in an area not associated with pressure-induced ulceration or venous stasis would raise suspicion of small-artery or arteriolar vasculitis.
Small-vessel vasculitis may also be associated with visceral involvement, including alveolar hemorrhage or glomerulonephritis. Visceral involvement most commonly occurs in Henoch-Schönlein purpura, cryoglobulinemia and vasculitis associated with autoimmune diseases, such as SLE. Extensive visceral involvement, however, should raise suspicion of coexistent medium-sized vessel disease.
VASCULITIS OF SMALL AND MEDIUM-SIZED VESSELS
Vasculitis of small and medium-sized vessels occurs in a heterogeneous group of disorders and includes the types of systemic vasculitis that are associated with the worst prognosis. No consensus exists on definitions for these syndromes, so the literature can be confusing.
Polyarteritis nodosa is the least distinctive of the vasculitides. Although polyarteritis nodosa is most often idiopathic, well-recognized associations include cryoglobulinemia, hairy cell leukemia, rheumatoid arthritis, Sjögren's syndrome and hepatitis B13; hence, the mnemonic “CLASH”—cryoglobulinemia, leukemia, arthritis, Sjögren's syndrome and hepatitis—is used to describe these associations. Patients with “classic” polyarteritis are often middle-aged and predominantly have medium-sized artery vasculitis. Although any organ system may be involved, the most common manifestations include peripheral neuropathy, mononeuritis multiplex, intestinal ischemia, renal ischemia, testicular pain and livedo reticularis.
Microscopic polyangiitis is a variant of polyarteritis and commonly presents as pulmonary hemorrhage and glomerulonephritis. Pathologically, polyarteritis shows focal, panmural infiltration and destruction of the vessel wall by neutrophils and, less commonly, by mononuclear cells, and eosinophils and fibrinoid necrosis. Thrombosis and aneurysms preferentially occur at sites of vessel bifurcation. In Wegener's granulomatosis, small and medium-sized arteries are affected by necrotizing granulomatous inflammation of the upper and lower respiratory tracts. Glomerulonephritis may also be present. This condition typically is manifested by recurrent sinusitis or epistaxis, mucosal ulcerations, otitis media, cough, hemoptysis and dyspnea.14 Destructive changes may lead to saddle-nose deformity or tracheal stenosis. Eye involvement may take the form of episcleritis, uveitis and proptosis related to orbital granulomas.
Churg-Strauss vasculitis occurs in the setting of allergic rhinitis, asthma and eosinophilia.13 Sinusitis and pulmonary infiltrates also occur but, in contrast to Wegener's granulomatosis, glomerulonephritis is uncommon, and cavitary lung nodules do not occur. Peripheral neuropathy, coronary arteritis and gastrointestinal involvement are common. Pathology shows prominent eosinophils and granulomas (Figure 3).
FIGURE 3.
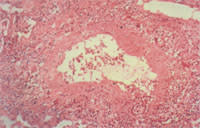
Specimen from an open lung biopsy in a 20-year-old patient with Churg-Strauss vasculitis. Extensive eosinophil-predominant inflammatory infiltrate surrounds and invades the small muscular artery.
Kawasaki disease (mucocutaneous lymph node syndrome) is an acute febrile illness that affects infants and small children.15 In this disease, necrotizing vasculitis involves small and medium-sized muscular arteries, with a predilection for the coronary arteries. Patients may present with fever, conjunctivitis, lymphadenopathy, erythematous desquamating rash, oral mucositis and arthritis. Myocarditis and coronary artery aneurysm formation may result in myocardial infarction early or late in the course of the disease.
VASCULITIS OF LARGE VESSELS
Giant cell, or temporal, arteritis is the most common primary vasculitis in the elderly. It characteristically affects the branches of the carotid arteries and causes headache. Other typical features include polymyalgia rheumatica, jaw or tongue claudication, scalp tenderness, constitutional symptoms and vision disturbances. Additionally, giant cell arteritis is a common cause of fever of unknown origin in the elderly.16 Patients with giant cell arteritis are almost always over age 50 and have an ESR over 50 mm per hour (“the rule of 50s”). The diagnosis is confirmed by temporal artery biopsy; in centers where this technique is available, the diagnostic yield may be enhanced if biopsy is performed under sonographic guidance that highlights potential areas of involvement. Irreversible blindness is the most common serious sequela of giant cell arteritis and is preventable with corticosteroid therapy.
Takayasu's arteritis affects the thoracic and abdominal aorta and its main branches or the pulmonary arteries.17 It primarily affects young women, particularly of Asian descent, and may be manifested by malaise, arthralgias and the gradual onset of extremity claudication. Most patients have asymmetrically reduced pulses, usually along with a blood pressure differential in the arms. Renal artery stenosis may occur. The diagnosis is made by arteriography.
Prognosis and Therapy
Although systemic vasculitis is a potentially life-threatening disorder, morbidity and mortality can be prevented if this disorder is recognized and treated early in its course. Initial therapy is primarily determined by the type and severity of organ involvement and by the rate of disease progression, although the specific type of vasculitis may further influence therapy.18
The use of high-dose corticosteroids (i.e., prednisone in a dosage of 1 mg per kg per day), occasionally in divided doses, is standard initial therapy for most of the systemic vasculitis syndromes. Corticosteroid therapy is often administered as intravenous “pulse” steroids (e.g., methylprednisolone in a dosage of 1 g per day for three days) in patients who have severe or life-threatening organ involvement. Immunosuppressive therapy, particularly cyclophosphamide (Cytoxan), azathioprine (Imuran) or methotrexate (Rheumatrex), in combination with corticosteroids, is widely used, but there are few supportive data from controlled, randomized trials.
This standard approach to treatment of systemic vasculitis may need to be modified, depending on the specific type of vasculitis. For example, the addition of oral cyclophosphamide to standard corticosteroid therapy provides a definite survival advantage in patients with Wegener's granulomatosis, and plasmapheresis can be life saving in patients with cryoglobulinemic crisis.
