Timely passage of the first stool is a hallmark of the well-being of the newborn infant. Failure of a full-term newborn to pass meconium in the first 24 hours may signal intestinal obstruction. Lower intestinal obstruction may be associated with disorders such as Hirschsprung's disease, anorectal malformations, meconium plug syndrome, small left colon syndrome, hypoganglionosis, neuronal intestinal dysplasia and megacystis-microcolon-intestinal hypoperistalsis syndrome. Radiologic studies are usually required to make the diagnosis. In addition, specific tests such as pelvic magnetic resonance imaging, anorectal manometry and rectal biopsy are helpful in the evaluation of newborns with failure to pass meconium.
The first stool is passed within 24 hours of birth in 99 percent of healthy full-term infants and within 48 hours in all healthy full-term infants.1 Failure of a full-term newborn to pass meconium within the first 24 hours should raise a suspicion of intestinal obstruction. Among premature infants, however, one study2 revealed that only 37 percent of 844 preterm infants passed their first stool in the first 24 hours; 32 percent had delayed passage of the first stool beyond 48 hours. In 99 percent of the preterm infants, the first stool was passed by the ninth day after birth.
Clinical Presentation of Meconium Retention
Failure to pass meconium combined with progressive abdominal distention, refusal to feed and vomiting of bilious intestinal contents are the classic clinical signs of intestinal obstruction in neonates. Abdominal examination often reveals distended loops of bowel, which may be visible or palpable. Anal inspection is essential to exclude the presence of anal atresia, perineal fistula with anal atresia, the membranous form of anal atresia and anal stenosis.
Plain radiographs of the abdomen do not allow differentiation of small bowel obstruction from large bowel obstruction. The differential diagnosis for small bowel obstruction in neonates includes duodenal atresia, malrotation and volvulus, jejunoileal atresia, meconium ileus and meconium peritonitis. Bilious vomiting, with or without abdominal distention, is usually the first sign of small bowel obstruction. The differential diagnosis for large bowel obstruction in neonates includes Hirschsprung's disease, anorectal malformations and meconium plug syndrome (Table 1).
TABLE 1 Differential Diagnosis of Conditions That May Be Associated with Failure to Pass Meconium in the Newborn
| Diagnosis | Frequency | Abnormal findings | Therapy |
|---|---|---|---|
| Hirschsprung's disease | 1/4,0003 | Tight anus, empty rectum, transition zone | Surgery |
| Meconium plug syndrome | 1/500 to 1/1,00010 | Meconium plugs | Rectal stimulation, enema |
| Meconium ileus | 1/2,80012 | Abdominal distention at birth, cystic fibrosis | Enema with intravenous fluids, surgery |
| Anorectal malformation | 1/4,000 to 1/8,00014 | Absent anus, tight anus or fistula | Dilatation, surgery |
| Small left colon syndrome | Rare | Transition zone* at splenic flexure | Enema, rarely, colostomy |
| Hypoganglionosis | Rare | Transition zone* | Medical, TPN, surgery |
| Neuronal intestinal dysplasia type A | Rare | Transition zone,* mucosal Inflammation | Medical, surgery |
| Neuronal intestinal dysplasia type B | Rare | Megacolon | Medical, rarely, surgery |
| Megacystis-microcolon-intestinal hypoperistalsis syndrome | Very rare | Microcolon, megacystis | TPN |
TPN = total parenteral nutrition.
*—Transition zone (from small- to large-diameter bowel) refers to radiographic visualization on contrast study.
In many cases of suspected neonatal intestinal obstruction, the clinical history and physical examination combined with plain abdominal radiographs, contrast enema radiographic examination, anorectal manometry and rectal biopsy eventually yield the diagnosis. The most difficult management decision is to decide between conservative management and emergency surgery. Ideally, all newborns suspected of having bowel obstruction should receive treatment at a center where a pediatric surgeon is available.
ILLUSTRATIVE CASE
A 3,480 g (7 lb, 7 oz) male infant was born after 40 weeks' gestation. There were no complications during the pregnancy and delivery. He did not pass meconium after birth, and he had the onset of vomiting on the first day. His abdomen became mildly distended. The infant was not able to feed, and abdominal distention increased.
Rectal examination revealed a tight anus. On the second day, flat and upright abdominal films demonstrated numerous loops of dilated bowel (Figure 1a). Barium enema radiographic examination showed that most of the dilated bowel was colon; no transition zone was seen (Figure 1b).
FIGURE 1A.
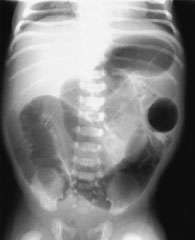
Plain abdominal radiograph demonstrating numerous loops of dilated bowel. Small bowel obstruction cannot be differentiated from large bowel obstruction. No gas is visible in the rectum.
FIGURE 1B.
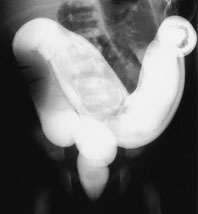
Barium enema radiograph showing an incompletely filled, dilated colon. This examination confirms that most of the dilated bowel on the plain film (Figure 1a) is the colon. No transition zone is visible.
The surgeon performed an anal dilatation, and the infant subsequently passed gas and meconium, which was followed by resolution of all symptoms.
After discharge from the hospital, the infant's mother continued performing periodic anal dilatation because he had difficulties moving his bowel. Digital rectal examination by the physician when the infant was five weeks of age revealed a tight anus and liquid stool but no impaction.
One week later, the mother noticed a bloody ring around his bowel movements. Barium enema radiographic examination at this time showed a transition zone in the distal portion of the sigmoid colon, with marked dilatation of the descending colon and left side of the transverse colon (Figure 1c). Anorectal manometry showed an absent rectosphincteric reflex. No ganglion cells were seen in the rectal biopsy. These findings were consistent with Hirschsprung's disease.
FIGURE 1C.
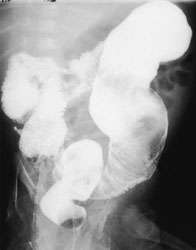
Barium enema radiograph performed when the infant in Figures 1a and 1b was six weeks of age, revealing a transition zone in the distal portion of the sigmoid colon, with marked dilatation of the descending colon and the left side of the transverse colon. These findings are consistent with Hirschsprung's disease.
Hirschsprung's Disease
Hirschsprung's disease, or congenital aganglionic megacolon, has an overall incidence of one in 4,000 live births.3 It accounts for 20 to 25 percent of the cases of neonatal bowel obstruction.4 The disease affects four times as many boys as girls, and 8 percent of patients with Hirschsprung's disease also have Down syndrome.5 The abnormal bowel innervation affects the internal anal sphincter. Most often, the rectosigmoid is involved, but a variable length of gut can be involved. A 30-year retrospective study6 revealed that the mean age at diagnosis has decreased to 2.6 months because of vigilance on the part of physicians, the use of anorectal manometry for assessment of the anal sphincter and early rectal biopsy to confirm the clinical diagnosis.
A common presentation of Hirschsprung's disease in the newborn is failure to pass meconium during the first few days of life, with subsequent passage of a meconium plug followed by sparse bowel movements. Gastrointestinal bleeding and diarrhea are danger signs for Hirschsprung's disease–associated enterocolitis. Enterocolitis can be fatal and is thought to be due to proliferation of bacteria as a result of stasis.
Physical examination often reveals the anus and rectum to be narrow and empty of stool. Plain abdominal radiographs show gas and stool in the colon and often the distention with stool or gas does not reach distally to the pelvic rim (Figure 1a).
Barium enema radiographic examination, performed with the colon unprepared, may reveal a transition zone that separates the small-to normal-diameter aganglionic bowel from the dilated bowel above (Figure 1c). A transition zone may not be recognizable in up to 25 percent of neonates with classic Hirschsprung's disease (Figure 1b). Similarly, a transition zone may not be discernible in patients with ultrashort segment Hirschsprung's disease, in patients with total colonic aganglionosis in whom the transition zone is above the colon and in patients who had an emergency colostomy. The presence of barium in the 24-hour delayed film is also suggestive of Hirschsprung's disease.
ANORECTAL MANOMETRY
When possible, anorectal manometry should be performed in all newborns with symptoms of lower bowel obstruction. With anorectal manometry, changes in anal pressure are recorded during and after rectal distention. When ganglion cells are present, rectal distention with a balloon inhibits the internal anal sphincter, resulting in a fall in anal pressure, called the rectosphincteric reflex (Figure 2a). In patients with Hirschsprung's disease, the rectosphincteric reflex is absent (Figure 2b).
FIGURE 2A.
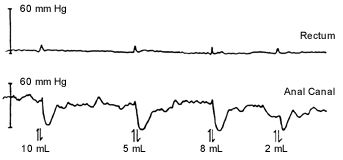
Anorectal manometric recording of a newborn with delayed passage of stool, showing a normal rectosphincteric reflex as demonstrated by the drop in anal pressure in response to rectal distention. The volume indicated is the volume used for rapid rectal distention. Rectal distention inhibits the internal anal sphincter, resulting in a fall in anal canal pressure.
FIGURE 2B.
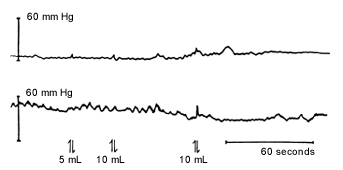
Anorectal manometric recording of a newborn with Hirschsprung's disease. The rectosphincteric reflex is absent, manifested by no change in the anal canal pressure in response to balloon dilation.
Anorectal manometry is most helpful in excluding the diagnosis of Hirschsprung's disease in a newborn.7,8 If the rectosphincteric reflex is absent, the diagnosis of Hirschsprung's disease needs to be confirmed by rectal suction biopsy, which shows no ganglion cells and markedly increased acetylcholinesterase staining of increased coarse neural fibers within the muscularis mucosae and the lamina propria.
TREATMENT
Surgery to remove or bypass the diseased bowel is required in all children with Hirschsprung's disease. In most neonates, a colostomy is initially placed into the normal bowel for decompression, followed by corrective surgery in three to six months. Occasionally, a primary pull-through procedure is performed.
Meconium Plug Syndrome
Meconium plug syndrome is the mildest and most common form of functional distal obstruction in the newborn. 9 It is a transient form of distal colonic or rectal obstruction caused by inspissated, immobile meconium. The incidence of meconium plug syndrome is estimated to range from one case in 500 to one case in 1,000 neonates.10 The etiology of this disorder is unclear.
The plain abdominal radiograph often reveals generalized gaseous distention of intestinal loops of small and large bowel filling the entire abdomen, but with no fluid levels. Contrast enema is diagnostic, showing the outline of the meconium plug (Figure 3), and also therapeutic if the plug is passed afterward. In some newborns, rectal stimulation with a thermometer, digital rectal examination or a saline enema induces passage of the plug. After the plug is passed, bowel movements are normal, and all symptoms resolve. However, even neonates with organic disease, such as Hirschsprung's disease, may pass a meconium plug and do well for a period of time. Therefore, continued observation is required in infants who pass a meconium plug and, if symptoms persist, further work-up is required.
FIGURE 3.
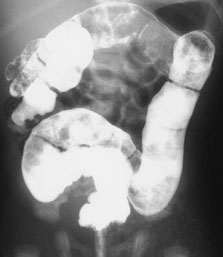
Barium radiograph of a newborn with meconium plug syndrome. Barium outlines multiple plugs of meconium. This infant passed the multiple plugs after the barium enema and then continued to pass normal meconium.
Meconium Ileus
Another cause of neonatal bowel obstruction by thick tenacious meconium is meconium ileus. Meconium ileus accounts for about 30 percent of cases of intestinal obstruction in newborns.11 In approximately 50 percent of the newborns with meconium ileus, the gut is undamaged and continuity is not disrupted; the obstruction is merely due to intraluminal meconium.9 In the other infants, meconium ileus is associated with volvulus, atresia or perforation.9 Cystic fibrosis is the underlying disorder in most infants with meconium ileus.9 Meconium ileus occurs in 15 percent of patients with cystic fibrosis. 12
Typically, abdominal distention is present at birth. Within hours, as air is swallowed, the distention increases, and the infant vomits bile-stained material. Thickened bowel loops are often palpable and visible through the abdominal wall. Massive distention, abdominal tenderness or abdominal erythema indicates the presence of complications. Rectal examination is often difficult because of the small caliber of the rectum.
Abdominal radiographs may reveal a distended bowel, few air-fluid levels and, in the right lower abdomen, meconium mixed with air, which has a ground-glass appearance on plain film. The presence of calcifications, free air or very large air-fluid levels suggests complications. The difference between meconium ileus and meconium plug syndrome is in the site and severity of the obstruction.
Contrast enema radiographic examination demonstrates a microcolon, often with no bowel contents. Reflux of contrast into the small bowel reveals the plugs. The small bowel is of narrow caliber below the plug and dilated above the plug.
Simple meconium ileus may be successfully treated by administration of a diatrizoate meglumine (Gastrografin) enema and plenty of intravenous fluids; the success rate is 16 percent12 to 50 percent.13 If the Gastrografin enema is unsuccessful, operative evacuation of the obstructing meconium by irrigation will be necessary. Complications such as atresia, perforation and meconium peritonitis always require immediate surgery, including resection, intestinal anastomosis and ileostomy.
Anorectal Malformations
One in 4,000 to one in 8,000 newborns is born with an anorectal malformation.14 A spectrum of anomalies of the lower intestinal tract and the genitourinary structures occurs from a failure of the completion of the complex embryologic developmental sequences in which the growth of the urorectal septum, lateral mesoderm structures and ectodermal structures form the normal rectum and lower urinary tract.
ANAL STENOSIS
Anal stenosis accounts for approximately 20 percent of anorectal malformations.4 The anus is very small, and a central black dot of meconium is present. Intense efforts are required to pass a ribbon-like stool. The diagnosis of anal stenosis is established by demonstration of a small, tight anus. Occasionally, an anal web may be the cause of the small anus. Anal dilatation is the usual treatment for anal stenosis and may need to be continued for several months.
ANAL ATRESIA
Anal atresia affects males and females with equal frequency. Perineal inspection reveals the absent anus (Figure 4). Broadly classified, anal atresia is characterized as “high” or “low,” depending on whether the rectum ends above the levator muscle or partially descends through this muscle. Often, the rectum ends in a fistula. In the high type of anal atresia, the fistula often ends in the prostatic urethra in males and in the vagina in females (Figure 5).
FIGURE 5.
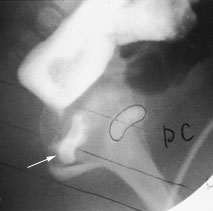
Barium enema radiograph showing evidence of imperforate anus and a vaginal fistula. Barium is present in the vagina (arrow). “PC” marks the pubococcygeal line.
Patients with a low type of anal atresia usually have a well-formed sacrum, a prominent midline groove and a prominent anal dimple. The low lesions are associated with a cutaneous fistula to the perineum. The orifice is small, located in the perineum anterior to the center of the external anal sphincter, close to the scrotum in the male and the vulva in the female. The abnormal anterior position of the fistula opening is occasionally not obvious and is mistaken for the anus. Males frequently have a black, ribbon-type structure in the perineum, which represents a subepithelial fistula filled with meconium.
Associated anomalies are present in up to 70 percent of patients with anorectal malformation.9 About 50 percent have urologic problems.15 The mnemonic VACTERL is used to describe the association of a combination of vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula with esophageal atresia, renal defects and radial upper limb hypoplasia.
Anal atresia requires surgical correction. The goal is to preserve bowel, urinary and sexual function. A colostomy is initially performed in neonates with high anal atresia. If a fistula is present on the perineum or in the vagina, it can be gently dilated to allow the gas and meconium to pass. Low lesions, including those with perineal fistulas, can be corrected electively when the infant's condition is stable.
Small Left Colon Syndrome
A rare cause of neonatal intestinal obstruction is small left colon syndrome, a functional distal bowel obstruction secondary to transient dysmotility in the descending colon.16 With this disorder, abdominal distention often develops after the newborn has passed meconium. More than 50 percent of newborns with small left colon syndrome are infants of mothers who have diabetes or an abnormal glucose tolerance test.16 Other infants are hypoglycemic or septic and, in others, an association with hypothyroidism, hypermagnesemia and maternal use of psychotropic drugs has been reported.
Plain radiograph shows dilated intestinal loops (Figure 6a) and, often, air-fluid levels. Contrast studies show the colon to be shortened and to lack the usual tortuosity from the anus to the splenic flexure. A sharp transition zone is seen at the splenic flexure (Figure 6b).
FIGURE 6A.
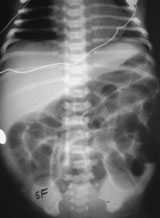
Plain radiograph of an infant with small left colon syndrome. Dilated intestinal loops are visualized.
FIGURE 6B.
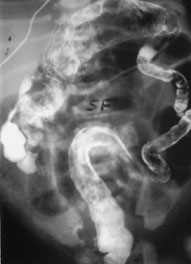
Rectal biopsy in patients with small left colon syndrome shows ganglion cells, contrary to the absence of ganglion cells in patients with Hirschsprung's disease. The ganglion cells can be normal, immature or hyperganglionic. The clinical course in newborns with small left colon syndrome varies in severity from mild symptoms, which may be relieved by the contrast enema, to severe bowel obstruction requiring a temporary transverse colostomy.16 Newborns with small left colon syndrome eventually have normal intestinal motility.
Other Causes of Failure to Pass Meconium
Various maternal medical conditions can cause a delay in meconium passage. In addition, maternal drug use, such as illicit drugs, magnesium sulfate and ganglionic blocking agents, can affect the infant and interfere with the passage of meconium. Neonatal medical conditions that can be associated with a failure to pass meconium include hypothyroidism, hypercalcemia, hypokalemia, sepsis and congestive heart failure.
Hypoganglionosis and neuronal intestinal dysplasia type A can produce symptoms and radiographic findings similar to those of Hirschsprung's disease.17–21 Both of these diseases are rare. They can affect part of or all of the gastrointestinal tract and sometimes occur in combination with Hirschsprung's disease. Histologically, hypoganglionosis is characterized by a reduced number of ganglion cells. In neuronal intestinal dysplasia type A, histologic features include hypoplasia or aplasia of the sympathetic innervation of the myenteric plexus and mucosa, along with mucosal inflammation.
Neuronal intestinal dysplasia type B may be manifested in the newborn period as meconium plug syndrome, small left colon syndrome or megacystis-microcolon-intestinal hypoperistalsis syndrome. The biopsy shows a dysplastic submucosal plexus and numerous giant ganglia, with many giant and small ganglion cells.
Various medications, partial or total parenteral nutrition, or surgery to remove or bypass segments of bowel may be necessary in patients with hypoganglionosis and neuronal intestinal dysplasia.
Another rare cause of neonatal intestinal obstruction is the megacystis-microcolon-intestinal hypoperistalsis syndrome.22 In this disorder, the small bowel is dilated and shortened, and the colon is a microcolon (Figure 7). There is an abundance of ganglion cells in the entire gastrointestinal tract. All patients with this syndrome have megacystis and megaureters, and most eventually die of complications from the disorder.
FIGURE 7.
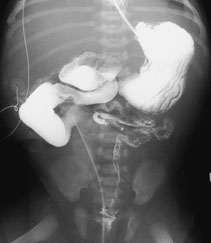
Barium enema radiograph in an infant with megacystis-microcolon-intestinal hypoperistalsis syndrome. The duodenum and proximal jejunum are mildly dilated. There was no significant progression of the contrast medium into the distal small bowel. The ligament of Treitz is to the right of the midline (malrotation). A colon series was performed three days before the barium enema, but contrast is still visible in the tortuous and abnormally positioned small colon. In addition, the urinary bladder is large and thickened, and reflux is present in the right dilated system.
