The symptomatic effects of drug abuse are a result of alterations in the functioning of the following neurotransmitters or their receptors: acetylcholine, dopamine, γ-aminobutyric acid, norepinephrine, opioids and serotonin. Anticholinergic drugs antagonize acetylcholine receptors. Dissociative drugs affect all transmitter sites. Opiates act on both opioid and adrenergic receptor sites. Psychedelic drugs stimulate serotonin release, and sedative-hypnotic drugs potentiate the γ-aminobutyric acid receptor. Specific signs and symptoms are associated with the neurotransmitters and receptors affected by each drug class. By recognizing symptomatic changes related to particular neurotransmitters and their receptors, family physicians can accurately determine the drug class and intervene appropriately to counteract drug-induced effects.
The symptoms of drug abuse are frequently misdiagnosed. The multiple signs and symptoms of intoxication and withdrawal often are not consistent because of variable dosages and the adulteration of drugs. These factors are further complicated by interactions related to multiple drug use, mixed intoxication-withdrawal states and idiosyncratic reactions. Misdiagnosis can result in significant morbidity and, sometimes, mortality.1
A biopsychiatric model may assist family physicians in the accurate diagnosis and efficient treatment of drug abuse. In this model, the signs and symptoms of drugs of abuse (Figure 1) are organized around the activity of six neurotransmitters. No known drug of abuse interacts singularly with the brain to produce unique symptom clusters. Rather, all known abused drugs affect a limited number of neurotransmitters by agonism or antagonism of a specific receptor site2 (Table 1).
FIGURE 1. Signs and Symptoms of Drug Intoxication and Withdrawal
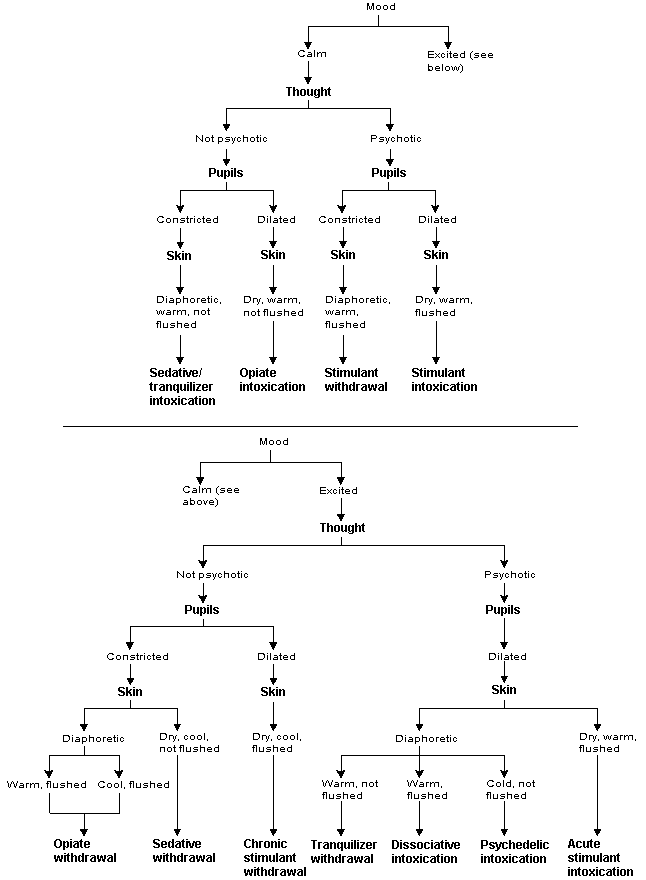
Proposed algorithm for the diagnosis of drug intoxication and withdrawal.
TABLE 1 Drug-Transmitter Actions That Cause Symptom Complexes
| Drug class | Neurotransmitters | |||||
|---|---|---|---|---|---|---|
| GABA | 5-HT | Norepinephrine | AcCH | β-Endorphin | Dopamine | |
| Opiates | X | X | ||||
| Dissociatives | X | X | X | X | X | |
| Psychedelics | X | |||||
| Stimulants | X | X | ||||
| Alcohol, sedatives, tranquilizers | X | X | X | X | ||
GABA = γ-aminobutyric acid; 5-HT = 5-hydroxytriptamine; AcCH = acetylcholine.
For clinical purposes, each receptor can be considered the site of action of only one specific neurotransmitter. Transmitters in the brain include acetylcholine, β-endorphin, dopamine, γ-aminobutyric acid (GABA), norepinephrine and serotonin. A knowledge of the symptoms associated with each neurotransmitter can facilitate diagnostic evaluation in drug abuse and withdrawal states (Table 2). As newer drugs of abuse are identified, physicians only need to know the affected neurotransmitter and receptor site to recognize the resultant signs and symptoms.
TABLE 2 Neurotransmitter Activity
| Neurotransmitter | Affected receptors | Clinical effects |
|---|---|---|
| Acetylcholine | Nicotinic and muscarinic receptors | Coordination, new memory, rapid-eye-movement sleep, affective expression, cognitive function |
| Dopamine | DA receptors | Cognitive integration, motor activity initiation, working memory |
| GABA | GABA receptors | Inhibitory transmission of cortex; functions of cerebellum, hippocampus and limbic system |
| Norepinephrine | α1, α2, β1 and β2 receptors | Sleep maintenance, mood modulation |
| Opioids | δ, κ ε, and μ receptors | Analgesia, euphoria, sedation, respiratory depression, hypothermia |
| Serotonin | All 5-HT receptors | Sleep initiation, mood modulation, pain modulation, modulation of aggression, control of anxiety, maintenance of alertness |
DA = dopamine; GABA = γ-aminobutyric acid; 5-HT = 5-hydroxytryptamine.
Each drug of abuse causes the release of one or more neurotransmitters. These neurotransmitters act on their respective neuroreceptor sites to produce the clinical effects of the particular drug.
The eight groups of abused drugs are anticholinergics, cannabinoids, dissociatives, opiates, psychedelics (hallucinogens), sedative-hypnotics, stimulants and volatiles (inhalants). Only six of these groups are important to family physicians involved in the treatment of intoxication, overdose and withdrawal states (Table 3). The exceptions are cannabinoids and volatiles.
TABLE 3 Drugs of Abuse: Six Groups That Are Likely to Require Primary Care Medical Intervention and Their Neurotransmitter Actions
| Drug class and members | Action on affected neurotransmitter | Neuroreceptors |
|---|---|---|
| Anticholinergics | Acetylcholine antagonists | Nicotinic and muscarinic |
| Asthmador | ||
| Benztropine (Cogentin) | ||
| Dimenhydrinate (Dramamine) | ||
| Diphenhydramine (Benadryl) | ||
| Hydroxyzine (Atarax) | ||
| Locoweed | ||
| Dissociatives | Affect actions of all neurotransmitters | All receptors |
| Ketamine (Ketalar) | ||
| Phencyclidine (PCP) | ||
| Phenylcyclohexylpyrolidine (PHP) | ||
| Opiates | β-Endorphin agonists | |
| Butorphanol (Stadol) | κ | |
| Pentazocine (Talwin) | ||
| Heroin | μ | |
| Hydromorphone (Dilaudid-Hp) | ||
| Mesipramine | ||
| Methadone | ||
| Morphine | ||
| Psychedelics | Serotonin agonists | 5-HT-2 |
| Borneol | ||
| Lysergic acid diethylamide (LSD) | ||
| Mescaline | ||
| Methylenedioxymeth-amphetamine (MDA) | ||
| Psilocybin | ||
| Sufrole | ||
| Sedative-hypnotics | GABA agonists | GABA-A |
| Barbiturates | ||
| Ethchlorvinyl (Placidyl) | ||
| Glutethimide (Doriden) | ||
| Methaqualone | ||
| Zolpidem (Ambien) | ||
| Benzodiazepines | GABA-A-α | |
| Ethyl alcohol | GABA and opioid agonist | GABA-A and μ |
| Stimulants | Dopamine, norepinephrine and serotonin agonists | DA-2, 5-HT-2, α and β |
| Amphetamine | ||
| Cocaine | ||
| Methamphetamine (Desoxyn) | ||
| Methylphenidate (Ritalin) |
GABA-A = subreceptor A of γ-aminobutyric acid-A; GABA-A-α = a-subsite of GABA-A; 5-HT-2 = subreceptor 2 of 5-hydroxytryptamine-1; DA-2 = subreceptor 2 of dopamine receptor.
Cannabinoids include Cannabis sativa products such as marijuana, hashish and hash oil. These products generally do not produce symptoms requiring clinical intervention. Volatiles such as glue, gasoline, formaldehyde, styrene and paint strippers can cause major damage to the brain, lungs, heart and liver. The effects of volatiles may require intensive inpatient intervention and are generally not managed by family physicians.
Commonly used drugs of abuse and their street names are listed by class in Table 4. Specific treatments for intoxication, overdose and withdrawal, based on the affected neurotransmitters, are presented in Table 5.
TABLE 4 Commonly Abused Drugs by Class
| Drug class and members | Street names | |
|---|---|---|
| Dissociatives | ||
| Ketamine (Ketalar) | K-hole, vitamin K | |
| Phencyclidine | PCP, angel dust | |
| Opiates | ||
| Fentanyl (Duragesic) | STP, six pack | |
| Heroin | China cat, skag | |
| Methadone | Orange barrel, dolphin | |
| Morphine | Morph | |
| Opium | Dust, yen shee | |
| Propoxyphene (Darvon) | Lilly | |
| Pentazocine (Talwin) | T's | |
| Aspirin-oxycodone (Percodan) | Perks | |
| Psychedelics | ||
| Lysergic acid diethylamide | LSD, blotter | |
| Mescaline | Aztec, blue cap | |
| Methylenedioxy-methamphetamine | Ecstasy, Adam | |
| Psilocybin | Purple passion, Aztec | |
| Sedative-hypnotics | ||
| Barbiturates | Ace, yellow jackets | |
| Ethchlorvinyl | Pickles, green jeans | |
| Glutethimide (Doriden) | CB, sen bee | |
| Meprobamate (Miltown) | Bams | |
| Methaqualone | Soapers, 714 | |
| Methyprylon | Roach 19, Easter bunny | |
| Rohypnol | Rib, date pill | |
| Alcohol | ||
| Beer | Colt, brew | |
| Distilled alcohol | Moon, scrap iron | |
| Wine | Mad dog, night train | |
| Benzodiazepines | Pumpkin seeds, roches | |
| Stimulants | ||
| Cocaine | Crack, coke | |
| Amphetamine | Hearts, speed | |
| Methamphetamine | Crank, crystal meth | |
| Methylphenidate (Ritalin) | White dragon, Ciba-19 | |
| Phenylpropanolamine (Propagest and others) | BT 72's, co-pilot | |
| Mixtures | ||
| Amphetamine/barbiturate | French blue | |
| Cocaine/heroin | Speed ball | |
| Cocaine/dissociative | Space base | |
| Opiate/marijuana | PJ | |
| Chloral hydrate/alcohol | Mickey Finn | |
| Pentazocine/antihistamine | T's & blues | |
| Phencyclidine/marijuana | Stepped-on grass | |
TABLE 5 Specific Treatment of Intoxication, Overdose and Withdrawal Based on Affected Neurotransmitter
| Neurotransmitter | Treatment |
|---|---|
| Intoxication and overdose | |
| Acetylcholine (anticholinergic) | Physostigmine (Antilirium) |
| β-Endorphin | Naloxone (Narcan) |
| Dopamine | Benzodiazepine |
| Butyrophenone | |
| GABA | Mechanical support |
| Norepinephrine | Beta blocker |
| Benzodiazepine | |
| Serotonin | Benzodiazepine |
| Withdrawal | |
| β-Endorphin | Methadone |
| Clonidine (Catapres) | |
| Dopamine | Bromocriptine (Parlodel) |
| GABA | Barbiturate or benzodiazepine replacement |
| Norepinephrine | Desipramine (Norpramin) |
| Serotonin | Fluoxetine (Prozac) |
GABA = γ-aminobutyric acid.
Anticholinergic Drugs
The anticholinergics are a diverse group of compounds with antagonism of cholinergic receptors as their unifying property. Because even small amounts can be fatal, anticholinergics constitute the most dangerous class of abused drugs.
Anticholinergic drugs act by antagonizing muscarinic and nicotinic cholinergic receptors. Muscarinic receptors are found in the reticular activating system, which maintains the waking state. Receptors in the basal ganglia are associated with major coordination, and receptors in the limbic system are associated with the expression of emotions. Receptors in the projection of the nucleus basalis are associated with memory formation and recall. Nicotinic receptors are located on sympathetic ganglia and voluntary muscles.
The classic presentation of anticholinergic overdose was described in about 1910. The anticholinergic drug abuser was characterized as “mad as a hatter, red as a beet, and dry as a bone.”3 This abuser usually complains of burning dysuria, dysphagia, constipation and diplopia. During higher levels of intoxication, hallucinations and body image distortions occur. The highest levels of intoxication can be life-threatening, producing delirium, coma, atonic bladder and cardiac arrhythmias.
The symptoms of anticholinergic overdose may be treated with physostigmine (Antilirium) in a dosage of 1 to 2 mg given intramuscularly or intravenously at 20-minute intervals. Physostigmine inhibits the acetylcholinesterase-induced breakdown of acetylcholine. This agent crosses the blood-brain barrier and thereby reverses both central and peripheral effects of the abused anticholinergic drug.
Some authorities have recommended against the use of physostigmine because of its frequent association with increased bronchial secretions and its rare association with asystole.4 However, when used correctly, this antidote can be a valuable treatment.5
Dissociative Drugs
Dissociatives act on all six neurotransmitter systems and produce a characteristic clinical presentation. Anticholinergic effects include dry, flushed skin and miosis. Stimulation of dopamine and norepinephrine release is responsible for rigidity, agitation, delusional thought, fever and excitement. Imperviousness to pain is mediated through the opioid systems. Perceptual changes in body image and hallucinations are apparently produced by action at the serotoninergic postsynaptic receptors. Excitatory activity is further enhanced by the inhibition of GABA receptors.
Because the dissociatives affect so many neurotransmitters, some discrimination is necessary in choosing a treatment protocol for intoxication. Most of the clinically significant symptoms of dissociatives are produced by presynaptic dopamine stimulation and cholinergic antagonism.5 These symptoms can be reversed with haloperidol (Haldol), which is a presynaptic dopamine antagonist. The dopamine-blocking action of haloperidol also shifts the dopamine-acetylcholine activity ratio in the limbic system. This “functionally” counteracts the anticholinergic actions of dissociatives.
Haloperidol is prescribed in an initial dosage of 5 mg given intramuscularly every 20 to 30 minutes until the patient is stabilized. Intramuscular administration of 1 g of ascorbic acid can potentiate the action of haloperidol and increase the rate of dissociative elimination by acidifying the urine.6
A second treatment option is risperidone (Risperdal). This agent counteracts the dopaminergic and serotoninergic effects of dissociatives but not their anticholinergic effects.7
Haloperidol has several advantages over risperidone. It can be administered intramuscularly, it works within 20 minutes and it is readily available in most emergency departments. In contrast, risperidone must be administered orally and takes one to two hours to work. Risperidone, however, is more useful in reducing paranoid behavior. It can also be useful when haloperidol produces no effect.7
Long-term use of dissociatives suppresses the production of norepinephrine and dopamine. Consequently, the abuser is likely to experience postwithdrawal depression. The depression can be treated with antidepressants that counteract this suppression. Desipramine (Norpramin), a relatively specific noradrenergic tricyclic antidepressant, is prescribed initially in a dosage of 50 mg taken at bedtime. The dosage is increased in 50-mg increments every other day to a final dosage of 200 mg taken at bedtime. This dosage is maintained for three to nine months.8
Opiates
Opiate-induced euphoria is mediated through the endogenous opioid systems in the brain. The opioid β-endorphin stimulates the μ receptors, thereby producing a sensation of well-being and providing general anesthesia. Opiates agonize the μ receptor, leading to repetitive use and subsequent tolerance and addiction. These drugs also inhibit the firing rate and release of norepinephrine in the locus caeruleus, which is a cell cluster in the pons.9
After repetitive opiate abuse, the β-endorphin system becomes functionally deficient. Consequently, opioid suppression of locus caeruleus activity is weakened. If the opiate is abruptly discontinued, the drug-induced euphoria is lost. This opiate withdrawal results in hyperactivity in the locus caeruleus, and subsequent increased noradrenergic release acts as a negative reinforcer of withdrawal.10
Two treatment approaches may be used to combat the symptoms of opiate withdrawal. The oldest approach involves substitution. A legal opiate analog such as methadone may be substituted for the abused opiate, with the methadone dosage then slowly reduced. A second approach is to disregard the opiate receptor and directly treat the withdrawal symptoms engendered by the norepinephrine “dump.” This is accomplished with clonidine (Catapres), an α2-adrenergic antagonist that reduces the rate of noradrenergic release and thereby attenuates the withdrawal symptoms. Although clonidine cannot prevent postwithdrawal depression, it has the advantage of not being addicting.11
Methadone is initially prescribed as a stabilization drug. First, the withdrawing addict is observed. If withdrawal symptoms develop to a level of great discomfort, methadone is prescribed. If the dosage of the abused drug is known, 1 mg of methadone can substitute for 2 to 4 mg of street heroin, 4 mg of morphine or 20 mg of meperidine (Demerol). If the level of abuse is not known, methadone is prescribed in a dosage of 10 to 15 mg per day taken orally. When appropriate and with support, the methadone dosage generally can be reduced by 20 percent per day.5
Clonidine is initially given in a dosage of 17 μg per kg per day divided into three or four doses. This dosage is maintained for several days and then gradually decreased.11
After detoxification, the patient can be referred to a drug rehabilitation clinic or Narcotics Anonymous.
If the patient feels that it is difficult to maintain a drug-free lifestyle, naltrexone (Trexan), an opiate receptor blocker, can also be used in maintenance therapy. Naltrexone produces nearly complete blockade of opioid receptors. As a result, it is nearly impossible to “get high” while taking naltrexone. The recovering opiate abuser generally takes a 25- to 50-mg oral dose of naltrexone in the morning. This dose blocks the effects of all opiate and opiate-like drugs for 24 or more hours.12,13 The only side effect of naltrexone is occasional depression.
Some family physicians prescribe naltrexone maintenance therapy for appropriate patients in their practices. However, methadone maintenance therapy requires supervision and thus is beyond the scope of most private practices and family medicine clinics.
Opiate intoxication generally does not require medical intervention. Naloxone (Narcan), a mixed opioid agonist–antagonist, may be given to treat opiate overdose. In most situations, 0.4 to 0.8 mg of naloxone is given intravenously every three to five minutes to a maximum dosage of 10 mg.14
Psychedelic Drugs
Psychedelic drugs, or hallucinogens, produce the equivalent of waking dreams. Some of these dreams may be pleasant; others may be nightmarish. The intoxicated state is characterized by illusions, visual hallucinations and bodily distortions. Physical signs usually include sweating, tachycardia and pupillary dilatation. These effects are thought to result from stimulation of serotoninergic activity in the pontine raphe nucleus, which produces disinhibition in the occipital lobe and limbic system. Because dopamine is not affected, reality testing remains intact.
Most psychedelic episodes remit within 12 hours and generally do not require medical treatment. Supportive care in a quiet environment with minimal visual stimulation is usually sufficient. The key is to reduce both internal and external stimuli that can amplify the serotonin-produced dreams. Both environmental and pharmacologic “screens” are effective.15
A pharmacologic screen can be produced by the indirect inhibitory effects of a benzodiazepine on serotonin release. If the patient becomes agitated or violent, the intramuscular administration of 1 to 2 mg of alprazolam (Xanax) should provide an adequate pharmacologic screen.
Psychedelic drugs are associated with no true withdrawal state. Discontinuance after chronic use, however, can produce a dysphoric state as a result of reduced serotonin activity. This dysphoric state can be relieved with fluoxetine (Prozac), a serotonin reuptake inhibitor. Fluoxetine is administered orally in a dosage of 20 to 40 mg per day for three to six months.16
Sedative-Hypnotic Drugs
Sedative-hypnotics (or simply sedatives) all act directly or indirectly on GABA receptors. These drugs fall into three subgroups: barbiturates and barbiturate-like drugs, minor tranquilizers and alcohol.
Barbiturates and barbiturate-like drugs such as phenobarbital and glutethimide (Doriden) potentiate the GABA-A receptor. The minor tranquilizers include benzodiazepines (e.g., alprazolam, diazepam [Valium]), halogenated benzodiazepines (e.g., temazepam [Restoril], flurazepam [Dalmane]) and zolpidem (Ambien). These drugs all increase the affinity of the α-subsite of the GABA binding site. Ethyl alcohol facilitates the effects of the GABA-A receptor but can also act directly on GABA-gated channels.17–19
Sedative-hypnotic overdose is generally not life-threatening unless a patient has taken more than one drug in this class or has added an opiate.
During sedative-hypnotic withdrawal, the regulatory function of GABA is diminished. This results in rebound of the previously suppressed stimulatory transmitters (serotonin, norepinephrine and dopamine). The withdrawal process must be carefully monitored because barbiturate or alcohol detoxification can be life-threatening.
In sedative-hypnotic detoxification, the patient is prescribed an equivalent dosage of phenobarbital (Table 6).5 Phenobarbital is preferred because it has a long half-life and thus does not have to be covered for its own subsequent withdrawal state. The duration of the detoxification program is determined by the drug that has been abused. The patient who has abused a short-acting sedative-hypnotic drug such as alprazolam or zolpidem can be detoxified in seven to 10 days, whereas the patient who has abused an intermediate-acting sedative-hypnotic such as diazepam, phenobarbital or glutethimide requires 10 to 14 days of detoxification.
During sedative-hypnotic withdrawal, blood pressure, body temperature and heart rate are monitored, and precautions should be taken to avoid the occurrence of seizures.5 Because of potential problems in these areas, detoxification is always conducted in an inpatient setting. The goal of detoxification is to reduce stimulant transmitter rebound. This is accomplished by agonizing the GABA receptors.
Nonintoxicated patients are given phenobarbital in a test dose of 60 mg taken orally. If no effect occurs, the patient is placed on an initial withdrawal schedule of 75 to 100 mg of phenobarbital taken orally four times daily. If the patient is alert with nystagmus, this initial dosage is reduced to 50 to 75 mg four times daily. The patient who is drowsy or manifests ataxia or slurred speech is initially treated with 30 to 50 mg of phenobarbital taken four times daily. The dosage of phenobarbital is reduced over a period of seven, 10 or 14 days, depending on whether the abused sedative is a short-acting, intermediate-acting or long-acting drug.
The withdrawal effects of benzodiazepines are less severe than those of the other sedative-hypnotics. Possibly this is due to indirect rather than direct action on GABA receptors through the subsite. Any benzodiazepine may be administered during benzodiazepine withdrawal.
First, the patient is observed. Treatment is initiated if the patient develops tremors, elevated body temperature, agitation or delirium. At six-hour intervals, the patient is given a benzodiazepine in a dosage equivalent to that of the benzodiazepine that has been abused (Table 7).5 Generally, a longer-acting benzodiazepine such as chlordiazepoxide (Librium) is used, and the initial dosage is titrated downward according to blood pressure elevation, pulse rate, temperature and psychotic symptoms. Withdrawal may be accomplished in three to seven days.
Physicians need to be aware that longer acting benzodiazepines may have a delayed manifestation of withdrawal. For example, the symptoms of diazepam withdrawal may not become evident until seven to 10 days after the drug is discontinued.20
Benzodiazepine intoxication requires no pharmacologic intervention.
Alcohol also affects the GABA receptors. At low doses, alcohol alters the GABA-A receptor's function; at higher doses, it can directly influence the opening of the chloride channel independently of GABA. Because of this action, alcohol produces similar effects as the sedatives but through an independent mechanism.
Alcohol intoxication rarely requires treatment, but it may precipitate seizures by lowering the seizure threshold level. Because the symptoms of alcohol withdrawal are related to a relative drop in alcohol levels, seizures may paradoxically occur during intoxication. These seizures are due to changes in the functioning of cell membranes, channels and transport systems caused by hypoglycemia, hypomagnesemia and increased intracellular sodium concentations.21
Alcohol withdrawal is treated similarly to benzodiazepine withdrawal. Generally, an effective starting dosage is 25 mg of chlordiazepoxide given orally three or four times daily or 1 mg of lorazepam given orally three or four times daily. Based on the symptoms, the dosage is gradually reduced over four to six days, and the drug is then discontinued.
Alternatively, the patient can be treated on an as-needed basis. With this approach, the patient is monitored, and chlordiazepoxide or lorazepam is given only when the patient has certain symptoms, such as gross resting tremors, visual hallucinations or a temperature greater than 38.3°C (101°F).22
Alcohol has some cross-reactivity with opioid receptors. This property can be exploited to reduce reinforcement of alcohol craving in the postwithdrawal state. Naltrexone, an opioid receptor antagonist, can be administered in a dosage of 25 to 50 mg per day.22
Stimulant Drugs
The stimulants promote the release of dopamine and norepinephrine from presynaptic neurons into the synaptic cleft but then block reuptake by these same neurons. Because of blocked reuptake, increased levels of catecholamines are available for receptor stimulation in the synaptic cleft.
One hypothesis is that progressive catecholamine depletion occurs because the catecholamines are exposed to synaptic metabolism and therefore are not available for reutilization by the presynaptic neurons.23 Thus, with each stimulant-induced catecholamine release-reuptake cycle, the catecholamine supply is slowly depleted. Recent research has shown that serotonin may also play a role.24–26
Bromocriptine (Parlodel), a dopamine receptor agonist, normalizes the effects of the dopamine depletion. Accordingly, bromocriptine is often used during stimulant withdrawal. This drug reduces stimulant craving and withdrawal symptoms. For stimulant detoxification, bromocriptine is given in an initial dosage of 0.625 to 2.5 mg taken orally three times daily. The dosage is then reduced by 0.625 mg per day over a period of three to 10 days.27
An alternative protocol uses desipramine, a tricyclic antidepressant. Desipramine produces a subsensitivity of dopamine and nor-epinephrine receptors. This presumably counteracts the increased dopaminergic and noradrenergic receptor binding that occurs in stimulant abuse. Consequently, desipramine has been reported to be effective in reducing the stimulant craving and postwithdrawal symptoms that occur because of the decreased catecholamine levels.
Desipramine may be used alone or with bromocriptine. The initial dosage of desipramine is 50 mg per day taken orally. This dosage is increased in 50-mg increments every other day until a dosage of 150 to 200 mg is achieved.26
Stimulant intoxication usually presents with hyperalertness and euphoria. Some addicts, especially chronic abusers, become paranoid or combative. The paranoia and combativeness generally remit without intervention after a few episodes. Increased release of catecholamines appears to be responsible for these changes.
If paranoia and combativeness become a problem or are associated with acutely threatening behavior, benzodiazepine therapy may be efficacious. The benzodiazepine indirectly reduces catecholaminergic activity by increasing GABA activity. The intramuscular administration of a 2-mg dose of lorazepam is usually sufficient.
Final Comment
Through careful observation of the specific signs and symptoms of drug intoxication, overdose or withdrawal, the affected neuro-transmitter(s) can be deduced. Knowledge of the affected neurotransmitter then dictates the appropriate pharmacologic intervention for overdose or withdrawal.
Whereas pharmacologic treatment of symptoms requires only a passive role on the part of the abuser, holistic treatment of the addict requires intense rehabilitation after withdrawal has been accomplished. Addiction is not a passive process. It requires an active decision to abuse, followed by repetitive voluntary episodes of drug use. After withdrawal, drug rehabilitation requires the active and voluntary involvement of the patient.
Pharmacologic intervention is only the first phase of the treatment process. The family physician must impress on the abusing patient the concept that addiction is a long-term, possibly lifelong, illness. The first intervention, detoxification, must be supplemented by inpatient and/or outpatient rehabilitation followed by regular participation in support groups such as Alcoholics Anonymous or Narcotics Anonymous. The patient must be directed to take responsibility for the disease of addiction and its treatment.28,29
