
Am Fam Physician. 2000;62(5):1119-1127
See related patient information handout on Cushing's syndrome and Cushing's disease, written by the authors of this article.
The most common endogenous cause of Cushing's syndrome is Cushing's disease. Frequent clinical findings include weight gain, truncal obesity, striae, hypertension, glucose intolerance and infections. Cranial nerve II may be affected by enlarging pituitary adenomas in Cushing's disease; cranial nerves III, IV and VI may also be affected. The evaluation of patients with suspected Cushing's disease and syndrome requires an understanding of the proper use and limitations of the tests commonly included in the diagnostic work-up. The best screening test for Cushing's syndrome is a 24-hour urine collection with analysis for urinary free cortisol excretion. Low-dose and high-dose dexamethasone suppression tests, corticotropin assays, a corticotropin-releasing hormone stimulation test and inferior petrosal sinus catheterization may be required for a definitive diagnosis. Magnetic resonance imaging is useful in localizing the lesion. Surgical removal of the lesion by a transphenoidal approach is usually successful, but long-term follow-up is required. Some patients require lifetime glucocorticoid replacement therapy.
The term “Cushing's syndrome” is used to describe a condition resulting from long-term exposure to excessive glucocorticoids. The syndrome is most commonly caused by the therapeutic administration of exogenous glucocorticoids.1 The term “Cushing's disease” is reserved for Cushing's syndrome that is caused by excessive secretion of adrenocorticotropin hormone (ACTH) by a pituitary tumor, usually an adenoma.2
Cushing's disease is responsible for roughly two thirds of the cases of endogenous Cushing's syndrome.1,3,4 The remainder of the endogenous cases are caused by ectopic ACTH-secreting tumors and primary adrenal neoplasms.5 Cushing's disease occurs most frequently in women of reproductive age, but it can affect males and females of any age.
The pituitary tumors in Cushing's disease are usually microadenomas, which, by definition, are 10 mm or less in diameter.3 Micro-adenomas generally do not cause symptoms by local mass effect. These tumors are most often discovered when clinical manifestations of hypercortisolism resulting from hypersecretion of ACTH prompt an appropriate diagnostic work-up. Occasionally, microadenomas are found incidentally during imaging performed for other reasons.
Macroadenomas are uncommon in patients with Cushing's disease. These tumors cause mass effect when their size exceeds 15 mm in diameter. Suprasellar extension and optic chiasm compression, local bone erosion, cavernous sinus compression and panhypopituitarism may occur as a macroadenoma enlarges.6
A common effect of elevated ACTH levels (whatever the source) is bilateral adrenocortical hyperplasia, which may be diffuse or nodular. Rarely, micronodular or macronodular ACTH-independent adrenal hyperplasia can be the cause of Cushing's syndrome.3
Presentations of Cushing's Disease
Patients with Cushing's disease usually present with one or more signs and symptoms secondary to the presence of excess cortisol or ACTH (Table 1). The clinical diagnosis must be based on the presence of one or more of these findings (Figure 1), because the syndrome itself has no true pathognomonic signs or symptoms.
| Sudden weight gain |
| Central obesity |
| Hypertension |
| Facial plethora |
| Proximal muscle weakness |
| Glucose intolerance or diabetes mellitus |
| Decreased libido or impotence |
| Depression or psychosis |
| Osteopenia or osteoporosis |
| Easy bruising |
| Hyperlipidemia |
| Menstrual disorders |
| Violaceous striae wider than 1 cm |
| Recurrent opportunistic or bacterial infections |
| Acne |
| Hirsutism |
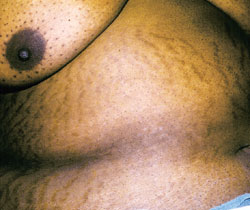
The most common symptom is sudden weight gain.3,6 Obesity, usually with a central distribution, is the most frequent sign. Any sign or symptom of cortisol excess can develop initially, but muscle weakness, bruising, hypertension, facial rounding and plethora eventually occur. Hypertension is likely to develop in patients who are more than 40 years of age.1
Pituitary tumors are known to compress the optic chiasm. Bitemporal hemianopsia with central visual field defects is the classic neuro-ophthalmologic finding, although other visual defects, including unilateral symptoms, can occur.7
Cranial nerve III (oculomotor nerve) palsies are less well recognized, but they may occur in up to 25 percent of patients with pituitary macroadenomas and may be the lone presenting symptom in patients with pituitary adenomas.8 Pituitary tumors infrequently affect cranial nerves IV and VI.9 The mechanism for oculomotor palsies is usually direct compression by the expanding tumor as the nerve passes through the walls of the adjacent cavernous sinus.7 Noncompressive cranial nerve III paresis has also been reported in patients with Cushing's disease.10
Another presentation or complication of Cushing's disease is opportunistic or bacterial infection. Immunosuppression resulting from corticosteroid excess is usually thought of as a cellular immune deficiency11 that increases the risk of opportunistic infections (Cryptococcus neoformans, Candida species, Norcardia species and other organisms).
Recent studies have also demonstrated the effect of glucocorticoids on the humoral immune system.12,13 By inhibiting activation of nuclear factor κ ß, glucocorticoids interfere with the production of a number of cytokines, including interleukin-6, which plays an integral role in mounting a response to bacterial infections.14
Pathophysiology of Cushing's Disease
When stimulated by ACTH, the adrenal gland secretes cortisol and other steroid hormones. ACTH is produced by the pituitary gland and released into the petrosal venous sinuses in response to stimulation by corticotropin-releasing hormone (CRH) from the hypothalamus (Figure 2). ACTH is released in a diurnal pattern that is independent of circulating cortisol levels: peak release occurs just before awakening, and ACTH levels then decline throughout the day. Control of CRH and ACTH release is maintained through negative feedback by cortisol at the hypothalamic and pituitary levels. Neuronal input at the hypothalamic level can also stimulate CRH release.
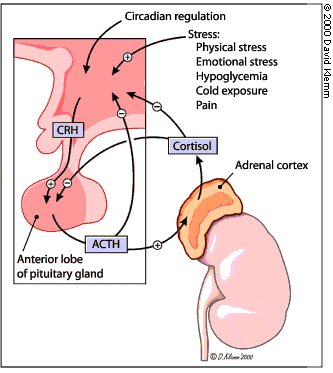
Although the adenomas of Cushing's disease secrete excessive amounts of ACTH, they generally retain some negative feedback responsiveness to high doses of glucocorticoids. Ectopic sources of ACTH, usually in the form of extracranial neoplasms, are generally not responsive to negative feedback with high doses of glucocorticoids. However, some overlap exists in the response to negative feedback between pituitary and ectopic sources of excessive ACTH.
Depression, alcoholism, medications, eating disorders and other conditions can cause mild clinical and laboratory findings, similar to those in Cushing's syndrome, termed “pseudo-Cushing's syndrome.” The laboratory and clinical findings of hypercortisolism disappear if the primary process is successfully treated.17–19
Dexamethasone, an exogenous glucocorticoid, is used to test for Cushing's syndrome. This gluococorticoid does not interfere with cortisol assays but induces similar physiologic responses.
Diagnostic Evaluation of Cushing's Disease
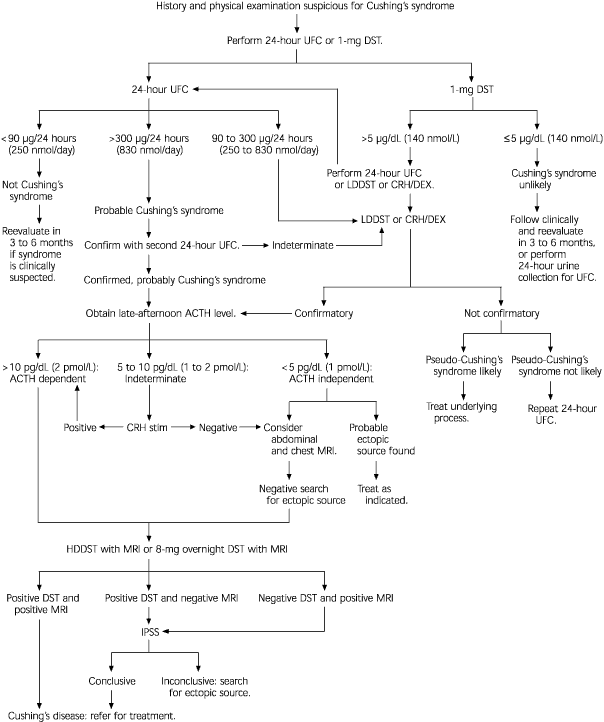
When Cushing's syndrome is suspected, initial laboratory testing is usually directed at confirming excessive glucocorticoid production. This is best accomplished through analysis of a 24-hour urine collection for urinary free cortisol excretion.4,20 Normal values are less than 90 μg per 24 hours (250 nmol per day). Values more than 300 μg per day (830 nmol per day) are considered diagnostic for Cushing's syndrome.4,5 The reported sensitivity of this test in detecting cortisol excess is 95 percent; the reported specificity is 98 percent.4 Ideally, the urinary free cortisol value should be confirmed with one or two additional measurements.
Because of the difficulty in obtaining 24-hour urine collections in many outpatients, some physicians use a l-mg overnight dexamethasone suppression test. For this test, the patient takes l mg of dexamethasone orally at 11 p.m., and the plasma cortisol level is measured at 8 a.m. the following day (normal value: 5 μg per dL or less [140 nmol per L]). The reported sensitivity of this test is 98 percent; the reported specificity is 80 percent.4
If the result of the dexamethasone suppression test is abnormal or the 24-hour urinary free cortisol level is mildly elevated, a confirmatory test for Cushing's syndrome is needed. The 24-hour urine collection for urinary free cortisol excretion can be used to confirm the result of the l-mg dexamethasone suppression test. Normal findings on both tests provide strong evidence against the presence of Cushing's syndrome.4 However, when Cushing's syndrome is still strongly suspected based on the clinical findings, negative tests should be repeated; the tests should also be performed again in three to six months.4
In the past, a low-dose dexamethasone suppression test was often performed to confirm the diagnosis of Cushing's syndrome. For this test, the patient takes 0.5 mg of dexamethasone orally every six hours for two days (a total of eight doses). A 24-hour urine collection is performed before the dexamethasone administration and again during the last 24 hours of dexamethasone administration. Patients with true Cushing's syndrome show less than 50 percent suppression of baseline urinary free cortisol excretion5 or more than 4 mg per 24 hours (11 μmol per day) of 17-hydroxysteroid excretion.4 This test is now used less often because of its low sensitivity (70 percent) and low specificity (75 percent).19
A newer approach is to combine a CRH stimulation test with a dexamethasone suppression test. The patient takes 0.5 mg of dexamethasone every six hours for two days (a total of eight doses); two hours after the last dexamethasone dose is taken, 1 μg per kg of CRH is administered intravenously. Blood for a plasma cortisol measurement is drawn 15 minutes after the CRH injection. A plasma cortisol level exceeding 1.4 μg per L (40 nmol per L) is considered positive for Cushing's syndrome.22 This test is nearly 100 percent sensitive and specific for Cushing's syndrome.4
Alternatively, if pseudo-Cushing's syndrome is suspected, a midnight serum cortisol level of less than 7.5 μg per dL (207 nmol per L) is strongly suggestive of pseudo-Cushing's syndrome.21
Another approach to the patient with suspected pseudo-Cushing's syndrome is to treat the underlying process and retest the patient in a few months.5 In this instance, the hypercortisolism of pseudo-Cushing's syndrome should correct spontaneously if treatment of the underlying process is successful.
Once hypersecretion of cortisol is confirmed, the next step is to determine whether the pathologic state is ACTH dependent or ACTH independent. This can be accomplished through measurement of the late-afternoon ACTH level. Late-afternoon (after 4 p.m.) timing is important because ACTH levels are normally low at that time.3 If the plasma ACTH level is greater than 10 pg per mL (2 pmol per L), the process is ACTH dependent. If the ACTH level is less than 5 pg per mL (1 pmol per L), the process is ACTH independent.4 Intermediate ACTH levels indicate the need for further study with, for example, a CRH stimulation test.3,4
The CRH stimulation test is performed by administering 1 μg per kg of CRH intravenously. ACTH and cortisol levels are measured before CRH injection and 15, 30, 45, 60, 90 and 120 minutes after injection. A rise in the cortisol value of 20 percent or more above basal level or a rise in the ACTH value of at least 50 percent above basal level is considered evidence for an ACTH-dependent lesion.4,5,23
When test results indicate that a patient has an ACTH-independent lesion, abdominal computed tomographic (CT) scanning or magnetic resonance imaging (MRI) is indicated to localize the site of the lesion.19
If the process is ACTH dependent, a high-dose dexamethasone suppression test combined with cranial MRI studies may aid in localizing the site of ACTH overproduction.
In the standard high-dose dexamethasone suppression test, a baseline 24-hour urine sample is collected. Then the patient takes 2 mg of dexamethasone every six hours for two days (total of eight doses). A 24-hour urine sample is collected during the last 24 hours of dexamethasone administration. The test is considered positive if 17-hydroxysteroid excretion is suppressed by 64 percent or urinary free cortisol excretion is suppressed by 90 percent of baseline. Using these criteria, the standard high-dose dexamethasone suppression test has been reported to be 86 percent accurate in differentiating Cushing's disease from ectopic ACTH production.3,4,19
Caution must be exercised in interpreting test findings. In patients with Cushing's disease, endogenous ACTH and cortisol production should be suppressed with high-dose (8-mg) dexamethasone, but the test lacks specificity, a clear cutoff value and diagnostic accuracy.3,6,19,24,25 In fact, the pretest probability of finding a pituitary cause for ACTH-dependent Cushing's syndrome (85 to 90 percent) may exceed the diagnostic accuracy of the test.4,25 Microadenomas are more likely to demonstrate high-dose dexamethasone suppression than macroadenomas (92 versus 56 percent in one study).26
An 8-mg overnight dexamethasone suppression test is easier to perform than the standard high-dose dexamethasone suppression test, and it has similar accuracy and limitations. In this test, a single 8-mg dose of dexamethasone is given at 11 p.m., and the serum cortisol level is measured at 8 a.m. the next morning. Suppression of the cortisol level to less than 50 percent of baseline is considered indicative of Cushing's disease.4,5,23
The CRH stimulation test can also help localize the site of ACTH overproduction. A 35 percent increase in the ACTH level at 15 and 30 minutes after corticotropin administration is reported to be 93 percent sensitive and 100 percent specific for Cushing's disease.4 The CRH stimulation test is considered to have a diagnostic accuracy similar to that of the high-dose dexamethasone suppression test, but it is more cost-effective because it can be performed in one morning in the outpatient setting.27
Some experts recommend using the combined results of the CRH stimulation test and the high-dose dexamethasone suppression test. A diagnostic accuracy of nearly 100 percent has been noted for the combined test results.28
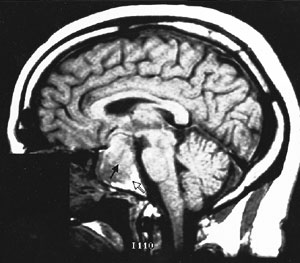
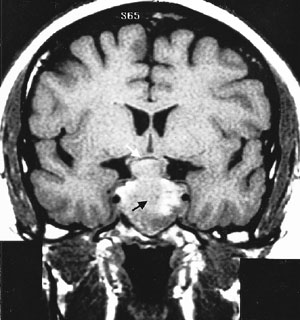
Cranial MRI results must also be interpreted cautiously. MRI may not visualize small lesions (Figures 4 and 5), and incidental lesions are noted on a small percentage of MRI scans in patients with ectopic ACTH production and no pituitary hyperfunction. Cranial CT scanning is not an acceptable substitute for MRI, because CT scans only identify approximately 50 percent of pituitary lesions.2
If the results of the work-up to this point are equivocal or the work-up suggests ectopic ACTH production, inferior petrosal sinus sampling is indicated.28 This procedure is considered the gold standard for definitive diagnosis of ACTH-dependent lesions.19 Inferior petrosal sinus sampling is invasive and expensive,3,19 but morbidity rates have been low in centers experienced in performing the procedure.4 In this technique, catheters are advanced into the petrosal venous sinuses bilaterally via the femoral vein. Samples are obtained from the periphery and petrosal sinuses simultaneously.
A petrosal sinus–to–peripheral ACTH ratio of 2:1 is considered diagnostic for a pituitary source.23,27 The diagnostic accuracy of the test exceeds 95 percent. The reported accuracy increases to 100 percent when a CRH stimulation test is added and the cutoff ratio is increased to 3:1.24 Inferior petrosal sinus sampling can also be used to localize the side of ACTH production from the pituitary gland.19
Treatment of Cushing's Disease
Transphenoidal removal of the tumor is the treatment of choice for Cushing's disease.29–31 Cure is likely if the patient develops hypocortisolism in the first few days to weeks after surgery.28,32 Most patients are rendered hypoadrenal for months to years after the procedure.33 During this period, they require glucocorticoid replacement therapy.
Ketoconazole (Nizoral) and aminoglutethimide (Cytadren), which are inhibitors of adrenal steroid biosynthesis, may be used in the perioperative period to decrease the clinical effects of hypercortisolism.3
Patients who have been surgically treated for Cushing's disease require careful long-term follow-up and monitoring for signs and symptoms of tumor recurrence. The pituitary adrenal axis must be evaluated six to 12 months after surgery to determine the potential need for lifetime exogenous steroid replacement therapy. Patients with panhypopituitarism subsequent to surgery require lifetime monitoring and titration of hormone therapy.
All patients who need glucocorticoid replacement therapy should be given careful instructions about the effects of stress and illness on glucocorticoid dosages. In addition, these patients should wear appropriate medical alert labels.
Final Comment
Patients with unexplained clinical findings suggestive of Cushing's syndrome should undergo a careful evaluation for hypercortisolism. If the presence of hypercortisolism is confirmed, its cause should be investigated. Cushing's disease is the most likely diagnosis in these patients.
In many instances, family physicians can perform the work-up of patients with suspected Cushing's disease and syndrome. Although definitive treatment of Cushing's disease requires a surgeon, family physicians have an important role in diagnosing the disease and providing continuing care after surgery.