Primary pulmonary hypertension is a rare disease of unknown etiology, whereas secondary pulmonary hypertension is a complication of many pulmonary, cardiac and extrathoracic conditions. Chronic obstructive pulmonary disease, left ventricular dysfunction and disorders associated with hypoxemia frequently result in pulmonary hypertension. Regardless of the etiology, unrelieved pulmonary hypertension can lead to right-sided heart failure. Signs and symptoms of pulmonary hypertension are often subtle and nonspecific. The diagnosis should be suspected in patients with increasing dyspnea on exertion and a known cause of pulmonary hypertension. Two-dimensional echocardiography with Doppler flow studies is the most useful imaging modality in patients with suspected pulmonary hypertension. If pulmonary hypertension is present, further evaluation may include assessment of oxygenation, pulmonary function testing, high-resolution computed tomography of the chest, ventilation-perfusion lung scanning and cardiac catheterization. Treatment with a continuous intravenous infusion of prostacyclin improves exercise capacity, quality of life, hemodynamics and long-term survival in patients with primary pulmonary hypertension. Management of secondary pulmonary hypertension includes correction of the underlying cause and reversal of hypoxemia. Lung transplantation remains an option for selected patients with pulmonary hypertension that does not respond to medical management.
Pulmonary hypertension is a complex problem characterized by nonspecific signs and symptoms and having multiple potential causes. It may be defined as a pulmonary artery systolic pressure greater than 30 mm Hg or a pulmonary artery mean pressure greater than 20 mm Hg.
The etiology of primary pulmonary hypertension is unknown. Secondary pulmonary hypertension can be a complication of many pulmonary, cardiac and extrathoracic conditions. Cor pulmonale is enlargement of the right ventricle as a consequence of disorders of the respiratory system. Pulmonary hypertension invariably precedes cor pulmonale. Unrelieved pulmonary hypertension, regardless of the underlying cause, leads to right ventricular failure.
Epidemiology
The estimated incidence of primary pulmonary hypertension is 1 to 2 cases per 1 million persons in the general population. During childhood, the condition affects both genders equally; after puberty, it is more common in women than in men (ratio: 1.7 to 1). Primary pulmonary hypertension is most prevalent in persons 20 to 40 years of age. The condition has no racial predilection.1
Secondary pulmonary hypertension is relatively common but is underdiagnosed. Reliable estimates of the prevalence of this condition are difficult to obtain because of the diversity of identifiable causes.
In persons more than 50 years of age, cor pulmonale, the consequence of untreated pulmonary hypertension, is the third most common cardiac disorder (after coronary and hypertensive heart disease).2
Pathophysiology
Normal pulmonary artery systolic pressure at rest is 18 to 25 mm Hg, with a mean pulmonary pressure ranging from 12 to 16 mm Hg. This low pressure is due to the large cross-sectional area of the pulmonary circulation, which results in low resistance. An increase in pulmonary vascular resistance or pulmonary blood flow results in pulmonary hypertension.
The World Health Organization (WHO) has proposed a classification system for pulmonary hypertension based on common clinical features (Table 1).3 Patients with pulmonary hypertension can also be classified according to their ability to function (Table 2).3
TABLE 1 World Health Organization's Diagnostic Classification of Pulmonary Hypertension
| Pulmonary arterial hypertension | ||
| Primary pulmonary hypertension | ||
| Sporadic disorder | ||
| Familial disorder | ||
| Related conditions | ||
| Collagen vascular disease | ||
| Congenital systemic-to-pulmonary shunt | ||
| Portal hypertension | ||
| Human immunodeficiency virus infection | ||
| Drugs and toxins | ||
| Anorectic agents (appetite suppressants) | ||
| Others | ||
| Persistent pulmonary hypertension of the newborn | ||
| Others | ||
| Pulmonary venous hypertension | ||
| Left-sided atrial or ventricular heart disease | ||
| Left-sided valvular heart disease | ||
| Extrinsic compression of central pulmonary veins | ||
| Fibrosing mediastinitis | ||
| Adenopathy and/or tumors | ||
| Pulmonary veno-occlusive disease | ||
| Others | ||
| Pulmonary hypertension associated with disorders of the respiratory system and/or hypoxemia | ||
| Chronic obstructive pulmonary disease | ||
| Interstitial lung disease | ||
| Sleep-disordered breathing | ||
| Alveolar hypoventilation disorders | ||
| Chronic exposure to high altitudes | ||
| Neonatal lung disease | ||
| Alveolar-capillary dysplasia | ||
| Others | ||
| Pulmonary hypertension resulting from chronic thrombotic and/or embolic disease | ||
| Thromboembolic obstruction of proximal pulmonary arteries | ||
| Obstruction of distal pulmonary arteries | ||
| Pulmonary embolism (thrombus, tumor, ova and/or parasites, foreign material) | ||
| In-situ thrombosis | ||
| Sickle cell disease | ||
| Pulmonary hypertension resulting from disorders directly affecting the pulmonary vasculature | ||
| Inflammatory conditions | ||
| Schistosomiasis | ||
| Sarcoidosis | ||
| Others | ||
| Pulmonary capillary hemangiomatosis | ||
Adapted with permission from Rich S, ed. Executive summary from the World Symposium on Primary Pulmonary Hypertension 1998, Evian, France, September 6–10,1998, cosponsored by the World Health Organization. Retrieved April 14,2000, from the World Wide Web:http://www.who.int/ncd/cvd/pph.html.
TABLE 2 Functional Assessment of Patients with Pulmonary Hypertension*
| Class I: | Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnea or fatigue, chest pain or near syncope. |
| Class II: | Patients with pulmonary hypertension resulting in slight limitation of physical activity. These patients are comfortable at rest, but ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope. |
| Class III: | Patients with pulmonary hypertension resulting in marked limitation of physical activity. These patients are comfortable at rest, but less than ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope. |
| Class IV: | Patients with pulmonary hypertension resulting in inability to perform any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnea and/or fatigue may be present at rest, and discomfort is increased by any physical activity. |
*—Modified from the New York Heart Association classification of patients with cardiac disease.
Adapted with permission from Rich S, ed. Executive summary from the World Symposium on Primary Pulmonary Hypertension 1998, Evian, France, September 6–10, 1998, cosponsored by the World Health Organization. Retrieved April 14,2000, from the World Wide Web: http://www.who.int/ncd/cvd/pph.html.
In primary pulmonary hypertension, the pulmonary vasculature is the exclusive target of disease, although the pathogenesis remains speculative. The most widely accepted theory suggests that certain persons may be predisposed to primary pulmonary hypertension. In these persons, various stimuli may initiate the development of pulmonary arteriopathy. Vascular-wall remodeling, vasoconstriction and thrombosis in situ all play a role.1
Collagen vascular disease,4 portal hypertension,5 human immunodeficiency virus (HIV) infection6 and anorectic agents7 may produce a clinical picture similar to that of primary pulmonary hypertension. The use of appetite-suppressant drugs for more than three months is associated with a greater than 30 times increased risk of developing pulmonary hypertension.8 In the United States, the anorexic agents fenfluramine and dexfenfluramine were recalled in September 1997, only 18 months after they were released. The WHO considers other appetite suppressants, such as amphetamines, to have a “very likely” causative role in pulmonary hypertension (Table 3).3
TABLE 3 Risk Factors for Primary Pulmonary Hypertension
| Drugs and toxins | |
| Definite causal relationship | |
| Aminorex | |
| Fenfluramine | |
| Dexfenfluramine | |
| Toxic rapeseed oil | |
| Very likely causal relationship | |
| Amphetamines | |
| Possible causal relationship | |
| Meta-amphetamines | |
| Cocaine | |
| Chemotherapeutic agents | |
| Unlikely causal relationship | |
| Antidepressants | |
| Oral contraceptives | |
| Estrogen therapy | |
| Cigarette smoking | |
| Demographic factors and medical conditions | |
| Definite causal relationship | |
| Gender | |
| Possible causal relationship | |
| Pregnancy | |
| Systemic hypertension | |
| Unlikely causal relationship | |
| Obesity | |
| Diseases | |
| Definite causal relationship | |
| Human immunodeficiency virus infection | |
| Very likely causal relationship | |
| Portal hypertension and/or liver disease | |
| Collagen vascular diseases | |
| Congenital systemic-to-pulmonary cardiac shunts | |
| Possible causal relationship | |
| Thyroid disorders | |
Adapted with permission from Rich S, ed. Executive summary from the World Symposium on Primary Pulmonary Hypertension 1998, Evian, France, September 6–10,1998, cosponsored by the World Health Organization. Retrieved April 14, 2000, from the World Wide Web: http://www.who.int/ncd/cvd/pph.html.
Pulmonary hypertension can be related to excessive pulmonary blood flow, such as occurs in congenital cardiac anomalies involving left to right shunts. When pulmonary blood flow is markedly increased and pulmonary vascular capacity is reached, any further increase in blood flow causes a rise in pressure.
Increased pulmonary pressure is also a potential consequence of any condition that impedes pulmonary venous drainage. The pulmonary hypertension that occurs in left ventricular dysfunction and mitral valve disease is the result of an increase in resistance to pulmonary venous drainage and backward transmission of the elevated left atrial pressure. More direct obstruction of pulmonary venous drainage occurs in association with unusual conditions such as mediastinal fibrosis and pulmonary veno-occlusive disease.
Pulmonary hypertension frequently occurs in response to alveolar hypoxia. A reduction in oxygen tension causes pulmonary vasoconstriction by a variety of actions on endothelium and smooth muscle. Chronic mountain sickness and sleep apnea9 are common etiologies of pulmonary hypertension associated with hypoxemia. Acidosis, which also causes pulmonary vasoconstriction, may compound the effects of hypoxia.10
Hypoxia-induced vasoconstriction and capillary obliteration occur in interstitial lung disease and chronic obstructive pulmonary disease (COPD), which is the most common cause of pulmonary hypertension. During acute exacerbations of COPD, hypoxia and uncompensated hypercarbia can increase pulmonary blood pressure.
Pulmonary hypertension may occur when blood flow through large pulmonary arteries is hindered. The classic cause is pulmonary embolism. Acute pulmonary emboli induce only a mild to moderate elevation of pulmonary artery pressure. Acutely, the right ventricle is unable to generate a systolic pressure greater than 50 mm Hg; a higher systolic value suggests a chronic process with right ventricular hypertrophy. Therefore, a massive pulmonary embolus may cause right ventricular failure but not severe pulmonary hypertension. Chronic thromboembolism can provoke severe pulmonary hypertension, but this condition occurs in fewer than 1 percent of patients with thromboembolic disease.11
Clinical Presentation
Pulmonary hypertension often presents with nonspecific symptoms (Table 4). These symptoms are often difficult to dissociate from those caused by a known underlying pulmonary or cardiac disorder. The most common symptoms—exertional dyspnea, fatigue and syncope—reflect an inability to increase cardiac output during activity. Typical angina may occur despite normal coronary arteries. The mechanism is unclear, but anginal chest pain may be due to pulmonary artery stretching or right ventricular ischemia.
TABLE 4 Symptoms and Signs of Pulmonary Hypertension
| Symptoms | Signs |
|---|---|
| Dyspnea on exertion | Jugular vein distention |
| Fatigue | Prominent right ventricular impulse |
| Syncope | Accentuated pulmonic valve component (P2) |
| Anginal chest pain | Right-sided third heart sound (S3) |
| Hemoptysis | Tricuspid insufficiency murmur |
| Raynaud's phenomenon | Hepatomegaly |
| Peripheral edema |
Hemoptysis resulting from the rupture of distended pulmonary vessels is a rare but potentially devastating event. Raynaud's phenomenon occurs in approximately 2 percent of patients with primary pulmonary hypertension but is more common in patients with pulmonary hypertension related to connective tissue disease.4 More specific symptoms may reflect the underlying cause of pulmonary hypertension.
Abnormalities detected on physical examination tend to be localized to the cardiovascular system. A careful examination often detects signs of pulmonary hypertension and right ventricular hypertrophy.
The findings on lung examination are nonspecific but may point to the underlying cause of pulmonary hypertension. For instance, wheezing may lead to a diagnosis of COPD, and basilar crackles may indicate the presence of interstitial lung disease.
Diagnostic Evaluation
A high index of suspicion, a meticulous history and a careful physical examination are paramount to the diagnosis of pulmonary hypertension. Particular attention should be given to previous medical conditions, drug use (legal and illegal) and family history. In addition, all systems should be carefully reviewed. Commonly, suspicion is increased by the presence of increasing dyspnea on exertion in a patient with a known cause of pulmonary hypertension.
In pulmonary hypertension, the electrocardiogram (ECG) may demonstrate signs of right ventricular hypertrophy, such as tall right precordial R waves, right axis deviation and right ventricular strain (Figure 1). The higher the pulmonary artery pressure, the more sensitive is the ECG.12 The chest radiograph is inferior to the ECG in detecting pulmonary hypertension, but it may show evidence of underlying lung disease12 (Figure 2). Not infrequently, recognition of pulmonary hypertension begins with the discovery of right ventricular hypertrophy on the ECG or prominent pulmonary arteries on the chest radiograph.
FIGURE 1
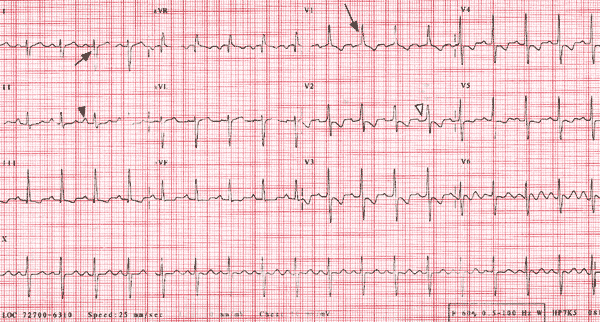
Electrocardiogram demonstrating the changes of right ventricular hypertrophy (long arrow) with strain in a patient with primary pulmonary hypertension. Right axis deviation (short arrow), increased P-wave amplitude in lead II (black arrowhead), and incomplete right bundle branch block (white arrowhead) are highly specific but lack sensitivity for the detection of right ventricular hypertrophy.12
FIGURE 2
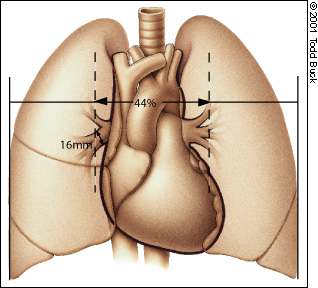
Anatomy of the thorax, along with indication of the upper limits of normal pulmonary vascular dimensions. A right interlobar pulmonary diameter of greater than 16 mm or a hilar-to-thoracic ratio of greater than 0.44 is specific but not sensitive for the diagnosis of pulmonary hypertension.12
Patients with signs, symptoms or electrocardiographic or radiographic findings suggestive of pulmonary hypertension should undergo two-dimensional echocardiography with Doppler flow studies. Echocardiography is the most useful imaging modality for detecting pulmonary hypertension13 and excluding underlying cardiac disease. Confirmation of pulmonary hypertension is based on identification of tricuspid regurgitation. The addition of mean right atrial pressure to the peak tricuspid jet velocity gives an accurate noninvasive estimate of peak pulmonary pressure. Right ventricular dilatation and hypertrophy are late findings.
All patients with documented pulmonary hypertension should undergo a comprehensive laboratory evaluation to clarify the etiology. The goal is to identify or exclude treatable causes. Initial tests include complete blood count, prothrombin time, partial thromboplastin time, hepatic profile and autoimmune panel (if this panel is suggested based on the history or physical examination). HIV testing should be considered in all patients, especially those with a compatible history or risk factors.
Arterial blood gas analysis should be performed to exclude hypoxia and acidosis as contributors to pulmonary hypertension. It is important to note that normal resting oxygenation does not exclude exertional or nocturnal oxygen desaturation. Approximately 20 percent of patients with COPD and normal awake arterial oxygen tensions have nocturnal nonapneic oxygen desaturation.14 Elevations of pulmonary artery pressure during transient oxygen desaturation are due to increases in pulmonary vascular resistance and cardiac output. These episodes are ameliorated with supplemental oxygen. Therefore, exercise and overnight oximetry should also be performed in all patients with pulmonary hypertension.
Pulmonary function tests are necessary to establish airflow obstruction or restrictive pulmonary pathology. Unless hypoxia is present, pulmonary hypertension cannot be attributed to these disorders until pulmonary function is severely reduced. Computed tomographic (CT) scanning of the chest with high-resolution images is useful for excluding occult interstitial lung disease and mediastinal fibrosis when the pulmonary function tests and chest radiograph are nondiagnostic.
If the cause of the pulmonary hypertension remains unexplained, chronic thromboembolism should be excluded before the diagnosis of primary pulmonary hypertension is accepted. Fortunately, ventilation-perfusion lung scanning is a reliable method for differentiating chronic thromboembolism from primary pulmonary hypertension. The finding of one or more segmental or larger perfusion defects is a sensitive marker of embolic obstruction. In primary pulmonary hypertension, the ventilation-perfusion scan is normal or demonstrates patchy subsegmental abnormalities.15
If the ventilation-perfusion scan suggests the presence of chronic thromboembolism, pulmonary angiography can be performed safely to confirm the diagnosis, define the extent of disease and evaluate the need for surgical thromboendarterectomy.16 The role of helical CT scanning of the pulmonary arteries remains unclear. This imaging technique has high specificity but undefined sensitivity for the diagnosis of pulmonary embolism.17
Cardiac catheterization should be performed in patients with unexplained pulmonary hypertension, and remains the gold standard for its diagnosis and quantification. Catheterization is particularly useful in diagnosing occult shunts, congenital heart disease and distal pulmonary artery stenosis.
An algorithm for the evaluation of suspected pulmonary hypertension is provided in Figure 3.
FIGURE 3 Evaluation of Suspected Pulmonary Hypertension
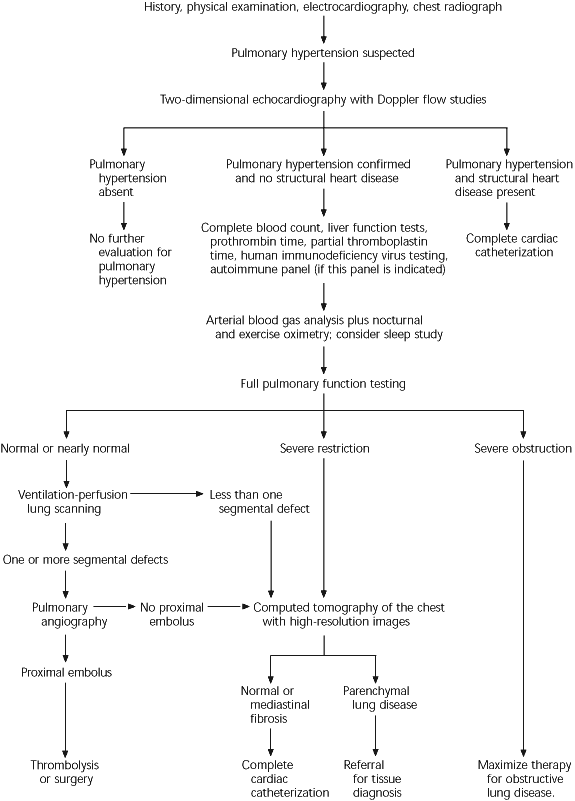
Algorithm for the evaluation of a patient with suspected pulmonary hypertension
Treatment
Some possible treatments for pulmonary hypertension are listed in Table 5. The treatment of primary pulmonary hypertension is complex, controversial and potentially dangerous.1 Patients benefit from referral to centers that specialize in the management of this uncommon problem.
TABLE 5 Possible Treatments for Pulmonary Hypertension
| Correct underlying cause: | |
| Surgical treatment of mitral stenosis, left to right shunt or accessible chronic thromboemboli | |
| Afterload reduction, digoxin (Lanoxin) and diuretics for left ventricular dysfunction | |
| Prevention and treatment of respiratory infection | |
| Avoidance of anorectic agents | |
| Decrease pulmonary vascular resistance: | |
| Vasodilators | |
| Oxygen | |
| Calcium channel blockers such as diltiazem (Cardizem) or nifedipine (Procardia) | |
| Prostacyclin (epoprostenol [Flolan]) or prostacyclin analogs | |
| Nitric oxide (investigational) | |
| Anticoagulants for primary pulmonary hypertension and chronic thromboembolism | |
| Increase cardiac output: | |
| Short-term parenteral inotropes | |
| Digoxin | |
| Reduce volume overload: | |
| Low-salt diet | |
| Diuretics | |
| Perform lung transplantation or atrial septosotomy (investigational) | |
Calcium channel blockers may alleviate pulmonary vasoconstriction and prolong life in about 20 percent of patients with primary pulmonary hypertension.18 Unfortunately, there is no way to predict which patients will respond to orally administered vasodilators, and these drugs frequently have significant adverse effects. Consequently, it is helpful to evaluate pulmonary vasoreactivity during catheterization, before a long-term therapy is selected. The most suitable drugs for testing acute response are potent, short-acting and titratable. In patients who show evidence of an acute hemodynamic response, long-term treatment with calcium channel blockers, administered orally in high dosages, can produce a sustained hemodynamic response and increase survival.18
Epoprostenol (Flolan), or prostacyclin, is the single most important advance in the treatment of primary pulmonary hypertension. This potent, short-acting vasodilator and inhibitor of platelet aggregation is produced by vascular endothelium. In one study,19 continuous intravenous infusion of epoprostenol improved exercise capacity, quality of life, hemodynamics and long-term survival in patients with class III or IV function (Table 2). Although the delivery system for continuous infusion is complex, most patients are able to learn how to prepare and infuse the drug.
Chronic anticoagulation with warfarin (Coumadin) is recommended to prevent thrombosis and has been shown to prolong life in patients with primary pulmonary hypertension.1 Patients with this condition are prone to thromboembolism because of sluggish pulmonary blood flow, dilated right heart chambers, venous insufficiency and relative physical inactivity. Maintaining an International Normalized Ratio of 1.5 to 2.0 is recommended. Other anticoagulants are also being studied.
Inotropic agents such as digoxin (Lanoxin) are currently under investigation. In one study,20 digoxin produced favorable acute hemodynamic effects in patients with right ventricular failure and primary pulmonary hypertension; however, the long-term consequences of this treatment are unknown. Shortterm parenterally administered inotropic drugs may also be of benefit.
In patients with secondary pulmonary hypertension, management is directed at early recognition and treatment of the underlying disease (while it is still potentially reversible). For instance, left ventricular dysfunction should be treated with afterloadreducing agents, digoxin and diuretics. Surgery to correct structural cardiac and pulmonary anomalies can also be effective, and thromboendarterectomy for accessible chronic thromboemboli is potentially curative.11 Improvement or resolution of pulmonary hypertension may occur after the discontinuation of anorectic agents, although resolution is not typical.21 Pulmonary hypertension associated with interstitial lung disease may respond to corticosteroids or other immunosuppressive agents.
Because hypoxia is a potent pulmonary vasoconstrictor, it is critical to identify and reverse hypoxemia. Low-flow supplemental oxygen therapy prolongs survival in hypoxemic patients.22 Failure to recognize and correct hypoxemia may be the error most frequently made in the treatment of patients with pulmonary hypertension.
A low-salt diet and judicious use of diuretics can be helpful in reducing volume overload in patients with pulmonary hypertension and right ventricular failure. Because the right heart is dependent on preload, care should be taken to avoid excessive diuresis and further reduction of cardiac output.
Patients with persistent pulmonary hypertension despite aggressive management of the underlying disease should be referred for evaluation at a center that specializes in the management of this condition. Unique protocols involving epoprostenol23 and other medications may be available to patients with secondary pulmonary hypertension that has not responded to more conventional measures.
Lung Transplantation
Primary pulmonary hypertension is usually progressive and ultimately fatal. Lung transplantation is an option in some patients younger than 65 years who have pulmonary hypertension that does not respond to medical management. According to a 1997 U.S. transplant registry report,24 lung transplant recipients with primary pulmonary hypertension had survival rates of 73 percent at one year, 55 percent at three years and 45 percent at five years. The immediate reduction in pulmonary artery pressure is associated with an improvement in right ventricular function. Recurrence of primary pulmonary hypertension after lung transplantation has not been reported.
Prognosis
The median duration of survival after the diagnosis of primary pulmonary hypertension is 2.8 years,25 but this figure is highly variable. As a result of new treatments, patients without hemodynamic evidence of right ventricular dysfunction may survive for more than 10 years.
The prognosis for patients with secondary pulmonary hypertension depends on the underlying disease, as well as right ventricular function. For instance, patients with COPD and moderate airflow obstruction have a three-year mortality rate of 50 percent after the onset of right ventricular failure.26 Survival is similarly influenced in patients with interstitial lung disease and pulmonary hypertension.
