Primary pulmonary hypertension (PPH) is familial in at least 6 percent of cases with an autosomal dominant inheritance, and is more common in women than in men. Anorexigens (such as fenfluramine and phentermine) have been associated with PPH in susceptible patients. In a department of JAMA entitled “Grand Rounds,” Gaine discusses the presentation, diagnosis and treatment of a patient with primary pulmonary hypertension.
Patients with PPH have progressive shortness of breath on exertion and, in more advanced disease stages, exertional chest pain, exertional dizziness, lightheadedness and overt syncope. Ortner syndrome (hoarseness resulting from compression of the left recurrent laryngeal nerve by an enlarged pulmonary artery) may be present, but cough and hemoptysis are uncommon. Initial physical findings include a loud pulmonic second sound (P2), indicating elevated pulmonary artery pressures. A bedside diagnosis of PPH can be made if the P2 is louder than the aortic second sound (A2) in the pulmonic area and remains louder as the stethoscope is moved down toward the apex of the heart. An audible right ventricular fourth heart sound (S4) is also common. A tricuspid regurgitant murmur may be heard, becoming more prominent as the right ventricle dilates. If the pulmonary valve ring also dilates, a Graham Steele pulmonary insufficiency murmur may result. In patients with advanced disease, a right ventricular third heart sound (S3) can be heard and is associated with a poor prognosis. A patient with advanced right heart failure will have jugular venous distention, edema and ascites. To meet the National Institutes of Health criteria for diagnosis of PPH, the mean pulmonary artery pressure must be greater than 25 mm Hg at rest or greater than 30 mm Hg in response to exercise.
A patient suspected of having PPH should be given certain diagnostic tests (see accompanying table), including a chest radiograph (may show enlarged pulmonary arteries but clear lungs); a ventilation-perfusion scan (may show mild abnormalities at the periphery); an echocardiogram (may show normal left heart function without defects in the septum); and pulmonary function tests (may show no airway obstruction but a mild reduction in lung volumes). Blood tests may be needed to exclude secondary causes of pulmonary hypertension, including antinuclear antibody, human immunodeficiency virus tests, liver function tests and hepatitis serology tests. A lung biopsy is necessary only in rare cases when the clinical diagnosis is uncertain.
Treatment of pulmonary hypertension is directed at the underlying etiology. For example, patients with emphysema should be treated with long-term oxygen therapy, bronchodilators and steroids to reduce the pulmonary artery pressure. Afterload reduction and diuretics are part of the therapy in patients with left ventricular dysfunction causing pulmonary venous disease. A patient with mitral valve disease may need valve repair or replacement to reduce the chronically elevated left atrial pressure. A patient with chronic thromboembolic pulmonary hypertension will need to be treated indefinitely with anticoagulation therapy, an inferior vena cava filter and a thromboendarterectomy to open the lumen.
Patients with pulmonary arterial hypertension usually have the following conditions: in situ thrombosis caused by endothelial dysfunction, smooth muscle hypertrophy and intimal and adventitial proliferation. Right heart catheterization confirms the diagnosis and helps to determine the most appropriate therapy when performed in conjunction with a vasodilator trial (see accompanying figure). Specific treatments that address each of these pathologic changes include warfarin to maintain an International Normalized Ratio between 1.7 and 2.2, vasodilators and continuous intravenous epoprostenol. The hemodynamic measurements (right atrial pressure, cardiac index and mean pulmonary artery pressure) obtained can predict a patient's life expectancy.
FIGURE. Pulmonary Hypertension
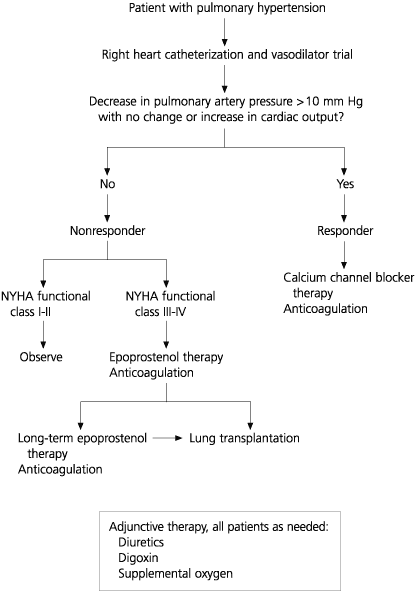
Treatment algorithm for pulmonary arterial hypertension. A right heart catheterization and vasodilator trial are performed to determine the approach to therapy. Patients who have a favorable acute response are treated with long-term calcium channel blocker therapy. Nonresponders and those who did not respond to calcium channel blockers are considered for continuous intravenous epoprostenol therapy either as a bridge to transplantation or as definitive long-term therapy. (NYHA = New York Heart Association)
Adapted with permission from Gaine S. Pulmonary hypertension. JAMA 2000; 284:3166.
A vasodilator trial (ideally using inhaled nitric oxide) can guide further treatment by determining whether the pulmonary artery pressure will respond to such agents. If the mean pulmonary artery pressure drops by greater than 10 mm Hg with a stable or increased cardiac output, the situation is considered reversible. In these patients, long-term oral vasodilator therapy with calcium channel blockers is recommended (see accompanying figure). In patients who are found to be nonresponders during the acute vasodilator trial, continuous intravenous epoprostenol therapy may be recommended, depending on their symptoms. Education of these patients on the management and administration of epoprostenol will need to be extensive. Oral and inhaled forms of epoprostenol-type medications are currently in clinical trials.
