Purpura is the result of hemorrhage into the skin or mucosal membrane. It may represent a relatively benign condition or herald the presence of a serious underlying disorder. Purpura may be secondary to thrombocytopenia, platelet dysfunction, coagulation factor deficiency or vascular defect. Investigation to confirm a diagnosis or to seek reassurance is important. Frequently, the diagnosis can be established on the basis of a careful history and physical examination, and a few key laboratory tests. Indicated tests include a complete blood cell count with platelet count, a peripheral blood smear, and prothrombin and activated partial thromboplastin times.
Purpura results from the extravasation of blood from the vasculature into the skin or mucous membranes. Therefore, purpuric lesions do not blanch with pressure. Depending on their size, purpuric lesions are traditionally classified as petechiae (pinpoint hemorrhages less than 2 mm in greatest diameter), purpura (2 mm to 1 cm) or ecchymoses (more than 1 cm).1 Although purpura itself is not dangerous, it may be the sign of an underlying life-threatening disorder. This article reviews the etiology of purpura in children and suggests an approach to evaluating the problem.
Normal Hemostasis
The normal hemostatic mechanisms are vascular response, platelet plug formation and activation of coagulation factors with the formation of fibrin to stabilize the platelet plug.
Following a vascular injury, vasoconstriction and retraction usually occur immediately and decrease blood flow to the affected area. Factor VIII–von Willebrand's factor (factor VIII–vWF) is released from endothelial cells and adheres to the exposed collagen matrix.2 Facilitated by factor VIII–vWF, platelets adhere to the endothelial cells of the damaged vessel wall and, in response to the exposed subendothelial collagen, release adenosine diphosphate (ADP) and thromboxane A2.3 The released ADP and thromboxane A2 cause further platelet aggregation and the formation of a platelet plug that is responsible for primary hemostasis.2,3
Secondary hemostatic mechanisms consist of a series of sequential enzymatic reactions involving various coagulation factors and leading to the formation of a fibrin clot. The intrinsic pathway is activated by the exposed collagen, and the extrinsic pathway is activated by tissue thromboplastin (Figure 1).3
The integrity of the vascular system depends on three interacting elements: platelets, plasma coagulation factors and blood vessels. All three elements are required for proper hemostasis, but the pattern of bleeding depends to some extent on the specific defect. In general, platelet disorders manifest petechiae, mucosal bleeding (wet purpura) or, rarely, central nervous system bleeding; coagulation disorders present as ecchymoses or hemarthrosis; and vasculitic disorders present with palpable purpura.1
Platelet Disorders
Simple purpura strongly indicates the presence of a qualitative or quantitative platelet disorder.
THROMBOCYTOPENIA
Thrombocytopenia may be caused by increased platelet destruction, decreased platelet production or sequestration of platelets.
Increased Platelet Destruction.
Immune thrombocytopenia is the most frequent cause of increased platelet destruction. Idiopathic (immune) thrombocytopenic purpura is by far the most common etiology of thrombocytopenia in childhood. The disorder is caused by the development of IgG autoantibodies to platelet membrane antigens as a result of an unbalanced response to an infectious agent or autoimmunity.4 The characteristic presentation is the sudden onset of bruises, purpura, mucosal hemorrhage and petechiae in a child who is otherwise in excellent health. An antecedent viral infection, especially an upper respiratory tract infection, is common. The peak incidence is between two and four years of age. Both genders are equally affected. Fever, lethargy, weight loss, bone pain, joint pain, pallor, lymphadenopathy and hepatosplenomegaly are characteristically absent. Minimal splenomegaly occurs in about 5 to 10 percent of symptomatic children.5 Idiopathic thrombocytopenic purpura is usually a temporary disorder, with 80 to 90 percent of children recovering within six to 12 months, usually within a few weeks.6 Chronic idiopathic thrombocytopenic purpura is more likely to present in teenage girls and children with underlying immune disorders. It has a more insidious onset.
FIGURE 1. Coagulation Cascade
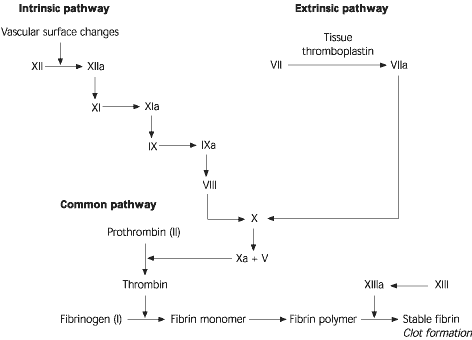
A simplified version of the coagulation “cascade.” An abnormality in the extrinsic pathway results in a prolonged prothrombin time (PT). An abnormality in the intrinsic pathway results in a prolonged activated partial thromboplastin time (aPTT). An abnormality in the common pathway results in prolongation of PT and aPTT.
Adapted with permission from Cohen AR. Rash—purpura. In: Fleisher GA, Ludwig S, et al., eds. Textbook of pediatric emergency medicine. 3d ed. Baltimore: Williams & Wilkins, 1993:430–8.
Drugs that act as a hapten with platelet surface antigens to form an immunologic moiety can also cause an immune thrombocytopenia. This has been shown to occur with penicillin, valproic acid (Depakene), quinidine, sulfonamides, cimetidine (Tagamet) and heparin.
Thrombocytopenia secondary to immune destruction may rarely be the presenting feature of human immunodeficiency virus, cytomegalovirus and herpesvirus infections.5 It also occurs in approximately 10 percent of patients with systemic lupus erythematosus; at times, thrombocytopenia may be the presenting manifestation.
Post-transfusion purpura is characterized by the acute onset of thrombocytopenia approximately five to 14 days after a transfusion.7 Previous transfusions may sensitize patients to foreign platelet antigens.
Neonatal isoimmune (alloimmune) thrombocytopenia develops when the mother produces alloantibodies in response to a fetal platelet antigen, most commonly P1A1, which is not present on maternal platelets. These IgG antibodies cross the placenta and cause thrombocytopenia in the fetus. The condition occurs most commonly in P1A1-negative women who have been previously sensitized to P1A1-positive platelets.2,5
Neonatal autoimmune thrombocytopenia may be caused by idiopathic thrombocytopenic purpura, systemic lupus erythematosus or drug-related immune-mediated purpura. In these disorders, maternal autoantibodies cross the placenta, bind to fetal platelets and cause their destruction.5
Nonimmune thrombocytopenia may occur because of hemolytic-uremic syndrome, thrombotic thrombocytopenic purpura or disseminated intravascular coagulopathy. Hemolytic-uremic syndrome is characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia and acute renal injury. The syndrome is most often associated with infection by verocytotoxin-producing Escherichia coli O157:H7. The pathologic process is initiated by toxin-induced endothelial injury and is followed by fibrin deposition in the renal microvasculature and destruction of red blood cells and platelets. In addition, vasoactive and platelet-aggregating substances are released from damaged endothelial cells and result in the formation of platelet thrombi.8
In hemolytic-uremic syndrome, thrombocytopenia may result from platelet destruction, increased consumption of platelets, intrarenal aggregation of platelets, sequestration of platelets in the liver or spleen, or a combination of these factors.8 The remaining platelets in the circulation appear to be “exhausted” and circulate in a degranulated form, depleted of nucleotide and granule contents. From a functional perspective, these platelets demonstrate a pattern characteristic of impaired aggregation.
Thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome have generally similar clinical and laboratory findings. However, thrombotic thrombocytopenic purpura occurs more often in adults, and neurologic (rather than renal) symptoms are more prominent.
Disseminated intravascular coagulopathy is characterized by generalized activation of the plasma coagulation pathways within small blood vessels, with the formation of fibrin and the depletion of all coagulation factors and platelets. This disorder may result from overwhelming sepsis, incompatible blood transfusion, snake bite, giant hemangioma and malignancy. Children with disseminated intravascular coagulopathy are generally quite ill and may present with extensive purpuric lesions and petechiae, as well as multifocal hemorrhage.9
Purpura fulminans is an acute, often lethal syndrome of disseminated intravascular coagulopathy. The skin lesions are rapidly progressive and characterized by microvascular thrombosis in the dermis, which ultimately results in perivascular hemorrhage and necrotic gangrene with minimal inflammation. Purpura fulminans may develop because of a severe bacterial infection, notably meningococcal disease, or because of protein C or S deficiency.10
Decreased Platelet Production.
A number of rare or uncommon congenital syndromes are associated with decreased platelet production.
Thrombocytopenia–absent radii (TAR) syndrome is inherited as an autosomal recessive trait. Purpura may present in the first few days of life or may be delayed for weeks.9
[ corrected] Fanconi anemia, also an autosomal recessive disorder, is characterized by pancytopenia, hyperpigmentation and café au lait spots, short stature, skeletal abnormalities and a wide array of integumentary and systemic abnormalities. Although the condition is congenital, hematologic abnormalities are not usually observed until the affected child is two to three years of age.9
Wiskott-Aldrich syndrome is characterized by microthrombocytopenia, eczema and recurrent infections secondary to immunodeficiency.9 The disorder is transmitted as an X-linked recessive trait. The thrombocytopenia results from abnormal platelet formation or release, despite quantitatively adequate numbers of megakaryocytes in the bone marrow.
Congenital amegakaryocytic thrombocytopenia is a rare cause of isolated thrombocytopenia in the neonatal period.9 Both autosomal dominant and recessive modes of inheritance have been described. In some children, the condition may be due to thrombopoietin deficiency.
Acquired causes of decreased platelet formation include drug reactions, infections and malignancies. Drugs such as alkylating agents, antimetabolites, anticonvulsants, chlorothiazide diuretics and estrogens can inhibit platelet production by suppressing megakaryocyte production.2,3
Thrombocytopenia resulting from bone marrow suppression is a common complication of viral and bacterial infections, especially septicemia. Intrauterine infection with TORCH organisms (toxoplasmosis, other [viruses], rubella, cytomegalovirus, herpes [simplex] viruses) may lead to thrombocytopenia in the neonatal period.9
Infiltration of bone marrow in patients with leukemia, histiocytosis, storage diseases, neuroblastoma, myelofibrosis and granulomatosis may result in thrombocytopenia. In osteopetrosis, the bone marrow space is replaced with frank bone formation.2
Sequestration of Platelets
Splenomegaly or giant hemangioma can result in thrombocytopenia because of platelet sequestration. Normally, approximately one third of the total platelet mass is in the spleen. Sequestration of platelets in an enlarged spleen, regardless of the cause, may lead to mild thrombocytopenia. Rarely, accelerated destruction of platelets may also occur.
The association of thrombocytopenia and giant hemangioma is referred to as Kasabach-Merritt syndrome.9 In addition to platelet trapping, consumption of coagulation factors and an increase in fibrin degradation products may also be present.
PLATELET DYSFUNCTION
Congenital Etiologies.
Glanzmann's thrombasthenia is an autosomal recessive disorder caused by congenital deficiency in the platelet membrane glycoproteins IIb and IIIa.9 The result is defective binding of platelet fibrinogen and a decrease in platelet aggregation with all stimulants except ristocetin.
Bernard-Soulier disease is an autosomal recessive disorder caused by a congenital deficiency in platelet membrane glycoprotein Ib and coagulation factors X and V.9 Affected patients have large platelets and decreased ristocetin-induced platelet aggregation.
Storage pool disease is a deficiency of dense granules, alpha granules, or both types of granules.9 Patients with storage pool disease have defective platelet release of ADP and serotonin.
Acquired Causes.
Drugs such as aspirin can cause inhibition of prostaglandin synthetase and thereby prevent the release of endogenous ADP and thromboxane A2, which are essential for platelet aggregation. Other drugs that may impair platelet function include furosemide (Lasix), nitrofurantoin (Furadantin), heparin, sympathetic blockers, clofibrate (Atromid-S) and some nonsteroidal anti-inflammatory drugs (NSAIDs).9
Reduced platelet adhesiveness has been demonstrated in uremic patients. Postulated causes include a nonspecific inhibitor or increased proteolytic activity in the circulation, which may enhance the catabolism of vWF. Deficiency of coagulation factors and a variety of abnormalities in platelet function have been described in patients with chronic liver disease. The impairment of fibrinolysis and the accumulation of fibrinogen degradation products can inhibit platelet aggregation.9
Coagulation Factor Deficiencies
Purpura can be the presenting symptom of congenital or acquired deficiency of coagulation factors.3
Hereditary deficiencies of virtually all coagulation factors have been described, but most are quite rare. The most commonly encountered disorders are coagulation factor VIII deficiency, coagulation factor IX deficiency and von Willebrand's disease. Coagulation factor VIII deficiency and coagulation factor IX deficiency (hemophilia type A and B) are inherited as an X-linked recessive trait. Spontaneous hemarthrosis is common in patients with these deficiencies but is rarely encountered in other coagulation factor deficiencies. Types I and II von Willebrand's disease are autosomal dominant, whereas type III is autosomal recessive. Type I disease is most common. Its severity is quite variable, and laboratory findings in the individual patient may vary over time.9
Acquired deficiencies of coagulation factors may be due to disseminated intravascular coagulopathy circulating anticoagulants, liver disease, vitamin K deficiency or uremia.3
Vascular Factors
CONGENITAL CAUSES
Hereditary hemorrhagic telangiectasia, an autosomal dominant disorder, is characterized by the development of fragile telangiectasia of the skin and mucous membranes. Because these blood vessels do not have sufficient muscle or connective tissue in their walls, hemostatic control is poor. Epistaxis and purpura are common presenting features.
Ehlers-Danlos syndrome, a group of genetically heterogenous connective tissue disorders, is characterized by skin hyperelasticity, joint hypermobility and fragility of the skin and blood vessels. Type IV is the ecchymotic form of the syndrome, and the collagen has a deficiency of hydroxylysine.9
ACQUIRED CAUSES
Acquired causes of vasculogenic purpura include Henoch-Schönlein purpura, infections, mechanical causes and psychogenic conditions.
Henoch-Schönlein purpura is an IgA-mediated systemic vasculitis of small blood vessels. The hallmarks are nonthrombocytopenic purpura, abdominal pain, arthritis and nephritis.11,12 This condition is the most common form of vasculitis in children.1 Approximately 75 percent of cases occur in children between two and 11 years of age.1 The condition is twice as prevalent in males as in females.11 From 60 to 75 percent of patients with Henoch-Schönlein purpura have a history of a preceding upper respiratory tract infection. Streptococcus is the most common infecting organism.13 Palpable purpura is present in almost 100 percent of patients with Henoch-Schönlein purpura. It is the presenting sign in 50 percent of patients.12 Some patients present with predominantly petechial lesions, some present with mainly purpuric lesions, and others present with a mixture of lesion types. Some patients have target-like lesions, with each lesion consisting of a central punctate hemorrhage surrounded by circumferential regions of pallor and hemorrhage.12 The rash is gravity and pressure dependent.14
TABLE 1 Findings of the History and Possible Etiologies of Purpura
| Historical data | Possible etiology |
|---|---|
| Age of onset | |
| Birth | Intrauterine infection, maternal idiopathic thrombocytopenic purpura, maternal systemic lupus erythematosus, maternal medication, TAR syndrome, congenital amegakaryocytic thrombocytopenia |
| 2 to 4 years | Idiopathic thrombocytopenic purpura |
| 4 to 7 years | Henoch-Schönlein purpura |
| Onset/chronicity | |
| Acute onset | Idiopathic thrombocytopenic purpura, Henoch-Schönlein purpura, medication, mechanical cause |
| Long duration | Abnormality of platelets, coagulopathy |
| Pattern of bleeding | |
| Mucosal bleeding | Thrombocytopenia, von Willebrand's disease |
| Intramuscular and intra-articular bleeding | Hemophilia |
| Associated symptoms | |
| Abdominal pain, blood in stools, joint pain | Henoch-Schönlein purpura |
| Lethargy, fever, bone pain | Leukemia |
| Intermittent fever, musculoskeletal symptoms | Systemic lupus erythematosus |
| Lethargy, polyuria, polydipsia, failure to thrive | Uremia |
| Purpura, but otherwise Healthy | Idiopathic thrombocytopenic Purpura |
| Drug use | |
| Alkylating agents | Thrombocytopenia |
| Antimetabolites | Thrombocytopenia |
| Past health | |
| Antecedent viral infection, especially an upper respiratory tract infection | Idiopathic thrombocytopenic purpura, Henoch-Schönlein purpura |
| Systemic lupus erythematosus | Systemic lupus erythematosus |
| Liver disease | Cirrhosis or chronic hepatitis |
| Renal disease | Chronic renal failure |
| Family history | |
| von Willebrand's disease | von Willebrand's disease |
| TAR syndrome | TAR syndrome |
| Wiskott-Aldrich syndrome | Wiskott-Aldrich syndrome |
| Maternal history | |
| Maternal idiopathic thrombocytopenic purpura | Immune thrombocytopenia |
| Maternal systemic lupus erythematosus | Immune thrombocytopenia |
TAR = thrombocytopenia-absent radii.
Drugs such as atropine and chloral hydrate may occasionally cause a purpuric lesion secondary to vascular involvement. This vascular purpura usually subsides when the drug is discontinued. Patients receiving long-term corticosteroid therapy may develop purpura secondary to defective vascular supportive tissue.
Mild transient nonthrombocytopenic purpura may result from measles, scarlet fever or typhoid. Meningococcemia and rickettsial diseases may cause direct damage to blood vessels, with resultant purpura.
Increased venous back pressure following violent coughing, vomiting or strangling may cause petechiae in the head and neck area. Linear lesions limited to readily accessible areas should raise the suspicion of factitious purpura. Some ecchymoses of bizarre shape are the result of religious rituals or cultural practices (e.g., cupping, coin rubbing or spoon scratching).15
Child abuse should be suspected if petechiae and bruises are widespread or found on areas of the body not normally subjected to injury. Typically, the platelet count and coagulation studies are normal in such children.5
Psychogenic purpura (also termed painful bruising syndrome, autoerythrocyte sensitization syndrome or Gardner-Diamond syndrome) is characterized by spontaneous painful ecchymoses in patients who often have psychologic instability.16
Clinical and Laboratory Evaluation
A thorough history (Table 1) and a careful physical examination (Table 2) are critical first steps in the evaluation of children with purpura. Bruises located only on the arms and legs of an active child are common findings and probably do not indicate a bleeding disorder. Careful observation, rather than exhaustive investigation, should be employed.
TABLE 2 Physical Findings and Possible Etiologies of Purpura
| Physical findings | Possible etiology |
|---|---|
| General findings | |
| Poor growth | Chronic disorder |
| Fever | Infection |
| Hypertension | Chronic renal failure, renal vasculitis |
| Characteristics of purpura | |
| Location on lower extremities | Henoch-Schönlein purpura |
| Location on palms and soles | Rickettsial infection |
| Palpable purpura | Vasculitis |
| Associated signs | |
| Arthritis, abdominal tenderness, subcutaneous edema, scrotal swelling | Henoch-Schönlein purpura |
| Hemarthrosis | Hemophilia |
| Butterfly rash, pallor, arthritis, lymphadenopathy | Systemic lupus erythematosus |
| Lymphadenopathy | Infection, drugs, malignancy |
| Jaundice, spider angioma, palmar erythema, hepatosplenomegaly | Liver disease |
| Pallor, lethargy, generalized bone tenderness, hepatosplenomegaly, lymphadenopathy | Leukemia |
| Skeletal anomalies | TAR syndrome, Fanconi's syndrome |
| Pallor, café au lait spots, short stature | Fanconi's syndrome |
| Telangiectasia | Hereditary hemorrhagic telangiectasia |
| Hyperelasticity of skin, hypermobility of joints | Ehlers-Danlos syndrome |
TAR = thrombocytopenia-absent radii.
When the history and physical examination suggest the presence of a bleeding disorder, laboratory screening studies may include a complete blood count, peripheral blood smear, prothrombin time (PT) and activated partial thromboplastin time (aPTT). With few exceptions, these studies should identify most hemostatic defects (Figure 2).3
A low hemoglobin level is indicative of blood loss, hemolysis or bone marrow failure. The presence of schistocytes points to hemolytic-uremic syndrome, thrombotic thrombocytopenic purpura or disseminated intravascular coagulopathy. An elevated reticulocyte count is found in hemolytic anemia.8
FIGURE 2. Diagnosis of Purpura
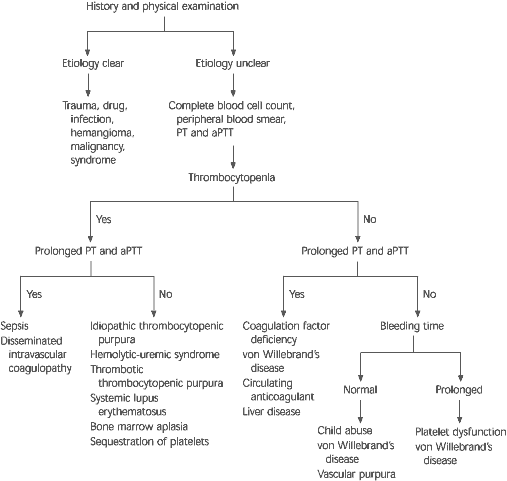
Algorithm for the diagnosis of purpura in children. (PT = prothrombin time; aPTT = activated partial thromboplastin time)
Adapted with permission from Cohen AR. Rash—purpura. In: Fleisher GA, LudwigS, et al., eds. Textbook of pediatric emergency medicine. 3d ed. Baltimore: Williams & Wilkins, 1993:430–8.
Neutrophilia and increased numbers of band forms or toxic granulations suggest a bacterial infection. Atypical lymphocytosis is seen in patients with infectious mononucleosis or cytomegalovirus infection. This finding may sometimes be confused with the blast cells of leukemia.
Anemia with thrombocytopenia indicates leukemia, systemic lupus erythematosus or aplastic anemia. If the platelet count is low but the rest of the complete blood count is normal, idiopathic thrombocytopenic purpura is the most likely diagnosis.
The mean platelet volume (MPV), now routinely reported by the automated cell counter (Coulter counter), can be of diagnostic significance. Macrothrombocytes (MPV greater than 10 fL) are seen in patients with idiopathic thrombocytopenic purpura, Bernard-Soulier disease or May-Hegglin anomaly, whereas microthrombocytes (MPV less than 6 fL) are seen in patients with aplastic anemia, Wiskott-Aldrich syndrome, TAR syndrome and some forms of storage pool disease.9
Bleeding time is a measure of the interval required for bleeding to stop after a standardized superficial incision is made on the forearm. This test is rarely indicated in children.
The PT is the time taken for citrated plasma to clot after the addition of tissue factor (thromboplastin) and calcium. A prolonged PT indicates a deficiency involving coagulation factors II, V, VII, X or fibrinogen.17 The aPTT is the time taken for citrated plasma preincubated with kaolin to clot after the addition of calcium and platelets. A prolonged aPTT is found in deficiencies involving coagulation factors II, V, VIII, IX, X, XI, XII or fibrinogen.
It should be remembered that an abnormal PT or aPTT only occurs when coagulation factor levels are less than 40 percent. Because of the immaturity of hepatic synthesis of coagulation factors, infants have physiologic prolongation of aPTT until they are three to four months of age. Patients with von Willebrand's disease often have mild and variable prolongation of PT and aPTT.3
Additional tests should be performed when indicated by the findings of the history, physical examination or screening laboratory tests. Measurements of coagulation factors (VIII, IX or vWF) are needed to confirm a specific diagnosis.
Platelet-associated antibodies may be present in patients with immune thrombocytopenia, but they are not sensitive enough for diagnostic purposes. If a platelet function defect is suspected, the following tests should be considered: platelet aggregation tests using activators (e.g., ADP, collagen, epinephrine, thrombin and/or ristocetin), clot retraction, prothrombin consumption test (for platelet factor III) and serotonin release.18
A bone marrow examination may be indicated if the cause of thrombocytopenia is not obvious, but it should definitely be performed if a second bone marrow cell line (i.e., red blood cells or white blood cells) is depressed. On the other hand, it has been shown that bone marrow aspiration rarely reveals an unexpected diagnosis when the clinical and laboratory findings are typical for idiopathic thrombocytopenic purpura.19
Hematuria may be present in Henoch-Schönlein purpura, systemic lupus erythematosus and hemolytic-uremic syndrome. Rheumatoid factor and antinuclear antibody tests should be ordered if a patient has significant prominent arthralgia or arthritis. Appropriate laboratory tests should be performed if kidney or liver failure is suspected.
Abdominal ultrasonography or computed tomographic scanning with contrast medium is appropriate when organomegaly is present. Cranial ultrasound examination is appropriate if the neonatal platelet count is less than 50 × 103 per μL (50 × 109 per L), even in the absence of neurologic abnormalities.
Management
The treatment of purpura should always be directed at its underlying cause. Specific treatment of the various causes of purpura is beyond the scope of this article but is discussed in standard pediatric hematology textbooks.9,20
Children with bleeding tendencies generally should not participate in strenuous activities or contact sports. In most instances, they should not receive intramuscular injections. The use of aspirin and other NSAIDs also should be avoided. Transfusion of platelets or coagulation factors may sometimes become necessary. Genetic counseling is useful in families with inherited bleeding disorders.
