Hereditary hemochromatosis is the most common inherited single-gene disorder in people of northern European descent. It is characterized by increased intestinal absorption of iron, with deposition of the iron in multiple organs. Previously, the classic description was combined diabetes mellitus, cutaneous hyperpigmentation and cirrhosis. Increasingly, however, hereditary hemochromatosis is being diagnosed at an earlier, less symptomatic stage. The diagnosis is based on a combination of clinical, laboratory and pathologic findings, including elevated serum transferrin saturation. Life expectancy is usually normal if phlebotomy is initiated before the development of cirrhosis or diabetes mellitus. Hereditary hemochromatosis is associated with mutations in the HFE gene. Between 60 and 93 percent of patients with the disorder are homozygous for a mutation designated C282Y. The HFE gene test is useful in confirming the diagnosis of hereditary hemochromatosis, screening adult family members of patients with HFE mutations and resolving ambiguities concerning iron overload.
Hereditary hemochromatosis is an autosomal recessive disorder associated with increased intestinal absorption of iron and deposition of excessive amounts of iron in the liver, pancreas, and other organs. It is the most common single-gene disorder in the U.S. white population. Approximately one in 250 to 300 white persons is homozygous for the hemochromatosis gene mutation, and at least one in 10 persons is a carrier for the mutation.1,2
Most physicians diagnose only a few cases of hereditary hemochromatosis in their practice because they do not routinely test for iron overload and because many patients with the disorder have no manifestations. It is estimated that the typical primary care physician encounters one patient with hereditary hemochromatosis every two weeks.
Iron overload caused by hereditary hemochromatosis should be distinguished from overload secondary to other entities. Secondary iron overload should be suspected in patients with chronic anemia, multiple transfusions, prolonged iron supplementation, or chronic liver disease. In secondary iron overload, iron often accumulates in Kupffer cells rather than hepatocytes, as typically occurs in hereditary hemochromatosis. However, severe iron overload from hereditary hemochromatosis or secondary causes may be indistinguishable. Disorders associated with iron overload are listed in Table 1.
TABLE 1 Disorders Associated with Iron Overload
| Hereditary hemochromatosis Related to HFE gene Not related to HFE gene
Thalassemia major Sideroblastic anemia Congenital dyserythropoietic anemia Congenital atransferrinemia | Exogenous iron overload Chronic iron supplementation (in absence of blood loss)
Viral hepatitis Alcoholic liver disease Nonalcoholic steatohepatitis Porphyria cutanea tarda Portacaval shunt |
Clinical Features
Persons with hereditary hemochromatosis absorb only a few milligrams of iron each day in excess of need. Therefore, clinical manifestations often occur only after 40 years of age, when body iron stores have reached 15 to 40 g (normally, the body stores approximately 4 g of iron). The relationship between total body iron stores and clinical manifestations over time is summarized in Figure 1.3
FIGURE 1. Iron Stores and Hereditary Hemochromatosis
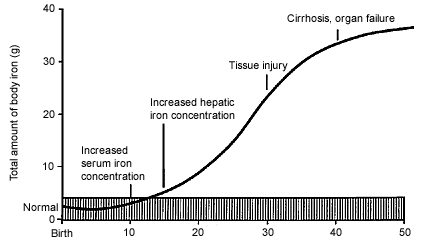
Relationship between total body iron stores and clinical manifestations of hereditary hemochromatosis over time.
Adapted with permission from Riely CA, Vera SR, Burrell MI, Koff RS. Inherited liver diseases. AGA clinical teaching project: unit 8. Bethesda, Md.: American Gastroenterological Association, 1993.
Disease expression may occur earlier in some persons and not at all in others. Clinical manifestation is influenced by age, sex, dietary iron, alcohol, blood loss in menstruation and pregnancy, and unknown factors. Although women are homozygous for the hemochromatosis mutation as often as men, they manifest the disease less frequently. Factors such as alcohol abuse and hepatitis C may accelerate disease expression.
In the past, hereditary hemochromatosis was usually diagnosed at an advanced stage. The classic description was cutaneous hyperpigmentation and diabetes mellitus in a patient with cirrhosis. Currently, most patients with newly diagnosed hereditary hemochromatosis are asymptomatic. The shift toward earlier diagnosis is probably the result of increased physician awareness and the inclusion of serum iron studies in many multichannel chemistry panels. Fatigue, arthralgias, and impotence are the most common symptoms of the disorder.4
Data on clinical features of hereditary hemochromatosis in patients diagnosed before and after 1990 are summarized in Table 2.5–9 If hereditary hemochromatosis is diagnosed early and treated appropriately, most (if not all) clinical manifestations are preventable. Once disease manifestations occur, however, many are irreversible (Table 3).
TABLE 3 Clinical Manifestations of Hereditary Hemochromatosis
| Reversible manifestations |
| Heart: cardiomyopathy, conduction disturbances |
| Liver: abdominal pain, elevated liver chemistries, hepatomegaly |
| Skin: bronzing (melanin deposition), gray pigmentation (iron deposition) |
| Infection:Vibrio vulnificus, Listeria monocytogenes, Pasteurella pseudotuberculosis |
| Irreversible manifestations |
| Liver: cirrhosis, hepatocellular carcinoma |
| Pituitary gland: gonadotropin insufficiency leading to secondary hypogonadism* |
| Pancreas: diabetes mellitus |
| Thyroid gland: hypothyroidism |
| Genitalia: primary hypogonadism |
| Joints: arthropathy in metacarpophalangeal joints, pseudogout |
*—Potentially reversible.
Diagnosis
The diagnosis of hereditary hemochromatosis is based on a combination of clinical, laboratory and pathologic criteria, including an elevated serum transferrin saturation and an elevated serum ferritin concentration. Serum transferrin saturation is calculated as follows:
Typical findings of iron studies in patients with hereditary hemochromatosis, along with normal reference ranges, are provided in Table 4.3 Because serum iron concentrations vary throughout the day and measurements may be affected by the ingestion of food, a test showing an elevated serum transferrin saturation should be repeated as a fasting early-morning determination. An elevated serum transferrin saturation is the earliest phenotypic abnormality in hereditary hemochromatosis.
TABLE 4 Iron Studies in Patients with Hereditary Hemochromatosis
| Iron study | Hemochromatosis | Normal range | |
|---|---|---|---|
| Serum iron concentration | 151 to 250 mcg per dL (27 to 45 μmol per L) | Men: 50 to 150 mcg per dL (9 to 27 μmol per L) Women: 35 to 145 mcg per dL (6 to 26 μmol per L) | |
| Total iron-binding capacity | 200 to 300 mcg per dL (36 to 54 μmol per L) | 250 to 400 mcg per dL (45 to 72 μmol per L) | |
| Serum transferrin saturation | 51% to 100% | 14% to 50% | |
| Serum ferritin concentration | |||
| Men | 300 to 6,000 ng per mL (300 to 6,000 mcg per L) | 20 to 300 ng per mL (20 to 300 mcg per L) | |
| Women | 200 to 6,000 ng per mL (200 to 6,000 mcg per L) | 20 to 200 ng per mL (20 to 200 mcg per L) | |
| Hepatic iron concentration (dry weight) | 5,000 to 30,000 mcg per g (89 to 550 μmol per g) | 100 to 2,200 mcg per g (5 to 27 μmol per g) | |
Adapted with permission from Riely CA, Vera SR, Burrell MI, Koff RS. Inherited liver diseases. AGA clinical teaching project: unit 8. Bethesda, Md.: American Gastroenterological Association, 1993.
Although the serum transferrin saturation is the best initial screening value, results may be normal early in the course of hereditary hemochromatosis. Furthermore, the serum ferritin concentration and serum transferrin saturation may be elevated in 30 to 50 percent of patients with acute or chronic viral hepatitis or alcoholic liver disease.
The serum ferritin concentration is a sensitive measure of iron overload, but it is also an acute-phase reactant and is therefore elevated in a variety of infectious and inflammatory conditions in the absence of iron overload. Consequently, it should not be used as the initial screening test to detect hereditary hemochromatosis.
Treatment
Treatment of hereditary hemochromatosis is usually reserved for patients with evidence of iron overload based on an elevated serum ferritin concentration. Phlebotomy is the preferred treatment because it is simple, effective and relatively inexpensive.
Phlebotomy entails removing 500 mL of blood on a weekly basis until the hemoglobin concentration, which is checked before each procedure, is lower than the reference range (approximately 12 to 13 g per dL [120 to 130 g per L]). Iron depletion is confirmed if the serum ferritin concentration is no higher than 50 ng per mL (50 mcg per L) and the serum transferrin saturation is less than 50 percent. Once iron depletion is accomplished, most patients require four to eight phlebotomies per year to keep the serum ferritin concentration lower than 50 ng per mL.
Patients with hereditary hemochromatosis should refrain from using iron supplements, including multivitamins that contain iron. A “low-iron diet” is not necessary, but red meat should be consumed in moderation. Patients with the disorder should avoid consuming (or even handling) raw seafood because of an associated increased risk of Vibrio vulnificus infection.10 They should also avoid or minimize alcohol use, because iron and alcohol are synergistic hepatotoxins.11
Genetic Testing
The gene associated with hereditary hemochromatosis is located on the short arm of chromosome 6. It was initially named HLA-H but has been renamed HFE.12 Two point mutations have been designated C282Y and H63D. More recently, other mutations have been described, but these are rare and are not likely to be of major clinical importance.13–17
In the initial study,12 as well as several other studies in the United States, Australia, and Europe, between 60 and 93 percent of patients with iron overload were found to be homozygous for the C282Y mutation.12,18 The wide range in prevalence of C282Y homozygotes may be due in part to different diagnostic criteria for hereditary hemochromatosis, as well as geographic differences in the prevalence of HFE mutations.19
The risk for iron overload is greatest in persons who are homozygous for the C282Y mutation. Iron overload also occurs in a minority of persons with other HFE mutations (especially compound heterozygotes, who have one copy of C282Y and one copy of H63D), but it is usually of lesser severity.
HFE gene testing may eliminate the need for liver biopsy in many patients. Traditionally, liver biopsy has been performed in patients with iron overload to confirm a diagnosis of hereditary hemochromatosis and to exclude cirrhosis. However, in patients with iron overload who are homozygous for the C282Y mutation, liver biopsy may be unnecessary to confirm the diagnosis of hereditary hemochromatosis.
Liver biopsy remains the “gold standard”for assessing the degree of fibrosis. Definitive exclusion of cirrhosis is important, because the risk of hepatocellular carcinoma is 200 times higher in patients with hemochromatosis and cirrhosis.8 The risk of cancer persists in these patients even after excess iron stores have been depleted. It may be appropriate to screen patients with cirrhosis every six months with an ultrasound examination and alphafetoprotein test.
Some patients with hereditary hemochromatosis have a minimal risk of developing cirrhosis and therefore may not require liver biopsy. A recent study20 confirmed that certain noninvasive predictors were accurate in excluding cirrhosis in patients who were homozygous for the C282Y mutation. In this study, no cases of cirrhosis occurred in 96 patients with the mutation who had serum ferritin concentrations lower than 1,000 ng per mL (1,000 mcg per L), normal aspartate aminotransferase values and no hepatomegaly.
The role of HFE mutation analysis in the diagnosis of iron overload disorders is summarized in Figure 2.21 The HFE gene test is most useful for screening adult family members of an identified proband. Screening family members is critical because 25 percent of the siblings and 5 percent of the children of a proband have hereditary hemochromatosis. HFE gene testing should replace the more cumbersome and expensive HLA typing previously used to screen siblings. In many instances, HFE gene testing is helpful for resolving ambiguous situations, such as iron overload associated with hepatitis C, alcoholic liver disease, and other causes of end-stage liver disease.
FIGURE 2. Screening for Hereditary Hemochromatosis
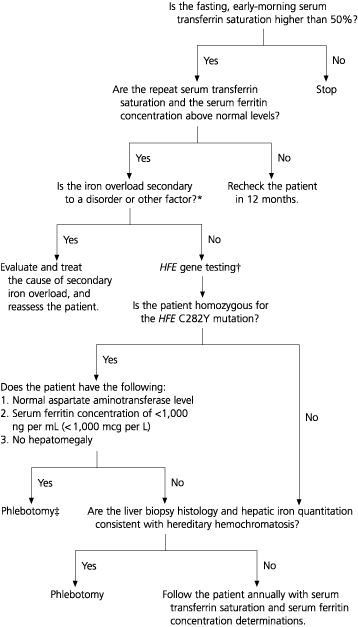
Suggested algorithm showing steps to follow in screening patients for hereditary hemochromatosis.
Adapted with permission from Brandhagen DJ, Fairbanks VF, Batts KP, Thibodeau SN. Update on hereditary hemochromatosis and the HFE gene. Mayo Clin Proc 1999;74:917–21.
Before an HFE gene test is performed, a qualified professional should provide counseling about the benefits and risks of genetic testing, as well as the alternatives to such testing. The possibility of insurance, employment, or other discrimination based on HFE test results is a concern. For this reason, HFE gene testing is usually not recommended for anyone younger than 18 years of age. This recommendation is consistent with the position of the National Institutes of Health Task Force on Genetic Testing.22
The HFE gene test is a polymerase chain reaction–based test that is usually performed on a whole-blood sample. The test is widely available at an average charge of approximately $200.
Screening
The issue of population screening for hemochromatosis was addressed at a 1999 international conference on hemochromatosis.23 At this conference, experts from various disciplines were unable to reach a consensus on whether to recommend population screening for hereditary hemochromatosis.
Hereditary hemochromatosis fulfills many criteria for a condition amenable to population screening. The disorder is common and has a long presymptomatic phase (Figure 1).3 Furthermore, a simple, noninvasive, inexpensive screening test exists, a simple, effective treatment is available, and treatment improves survival24 (Figure 3).8
FIGURE 3. Survival in Hereditary Hemochromatosis
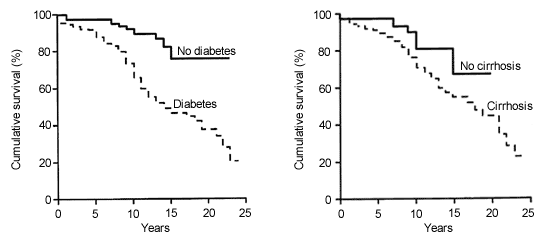
Cumulative survival in patients who have hereditary hemochromatosis with and without cirrhosis and diabetes. Survival in patients without diabetes or cirrhosis is similar to that in an age- and sex-matched control population without hemochromatosis.
Adapted with permission from Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch HJ, Strohmeyer G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med 1985;313:1256–62.
The World Health Organization (WHO) has established criteria to evaluate population screening for a medical condition.25 Recently, one investigator25 applied the WHO criteria to hereditary hemochromatosis and concluded that the disorder is appropriate for population screening. Using reasonable estimates of the number of patients who will develop life-threatening disease, investigators in several other studies26–28 concluded that population screening for hemochromatosis would be cost effective.
Although hereditary hemochromatosis fulfills many of the criteria for population screening, the U.S. Preventive Services Task Force and other organizations have not recommended screening for this disorder. Some public health experts29,30 cite lack of information about disease burden and expression as reasons for not endorsing population screening.
The natural history of hereditary hemochromatosis in an asymptomatic patient identified by population screening is unknown and may never be known because it would be unethical to withhold treatment once a patient develops iron overload. Another area of uncertainty is the proportion of persons homozygous for the C282Y mutation who will develop clinically significant problems with iron overload. Without this information, it will be difficult to obtain estimates on the cost-effectiveness of screening for hereditary hemochromatosis in the general population.
Five studies1,2,31–33 have compared HFE genotypes with serum transferrin saturation and serum ferritin concentration in the general population. In these studies, 19 to 75 percent of the subjects who were homozygous for the C282Y mutation had an elevated serum ferritin concentration, and 64 to 100 percent had an elevated serum transferrin saturation. The results of these studies are summarized in Table 5.1,2,31–34
Large population screening studies are currently in progress in more than 15 countries. A population screening study of 100,000 people in the United States and Canada was scheduled to begin in February 2001. It is hoped that these studies will provide more information about disease expression in hereditary hemochromatosis and help to determine whether screening should be considered in the general population. With the information currently available, it is reasonable to screen for hereditary hemochromatosis in patients who have chronic liver disease, signs or symptoms associated with hereditary hemochromatosis, or a family history of iron overload.