Bullous dermatoses can be debilitating and possibly fatal. A selection of autoimmune blistering diseases, including pemphigus vulgaris, paraneoplastic pemphigus, bullous pemphigoid, cicatricial pemphigoid, dermatitis herpetiformis and linear IgA dermatosis are reviewed. Pemphigus vulgaris usually starts in the oral mucosa followed by blistering of the skin, which is often painful. Paraneoplastic pemphigus is associated with neoplasms, most commonly of lymphoid tissue, but also Waldenström's macroglobulinemia, sarcomas, thymomas and Castleman's disease. Bullous pemphigoid is characterized by large, tense bullae, but may begin as an urticarial eruption. Cicatricial (scarring) pemphigoid presents with severe, erosive lesions of the mucous membranes with skin involvement in one third of patients focused around the head and upper trunk. Dermatitis herpetiformis is intensely pruritic and chronic, characterized by papulovesicles and urticarial wheals on the extensor surfaces in a grouped or herpetiform, symmetric distribution. Linear IgA dermatosis is clinically similar to dermatitis herpetiformis, but it is not associated with gluten-sensitive enteropathy as is dermatitis herpetiformis.
There is a wide variety of blistering diseases, some of which can be extremely debilitating and even fatal. Many of these diseases are autoimmune in nature (Table 1) and may also be associated with certain human leukocyte antigen types. Some bullous diseases have serious sequelae, necessitating early treatment and intervention to prevent further morbidity or mortality. Auto-immune blistering diseases include pemphigus vulgaris, paraneoplastic pemphigus, bullous pemphigoid, cicatricial pemphigoid, dermatitis herpetiformis, and linear IgA dermatosis (Table 2).
TABLE 1 Differential Diagnosis of Autoimmune Bullous Dermatoses
| Pemphigus vulgaris |
| Paraneoplastic pemphigus |
| Bullous pemphigoid |
| Cicatricial pemphigoid |
| Dermatitis herpetiformis |
TABLE 2. Summary of Autoimmune Bullous Dermatoses
| Disorder | Epidemiology | Clinical features | Diagnosis | Treatment | Comments |
|---|---|---|---|---|---|
| Pemphigus vulgaris | Rare, equal in men and women, onset at 40 to 60 years of age, more common in Jewish or Mediterranean descendants | Flaccid blisters or crushed erosions, located on head, upper trunk, intertriginous areas, and mucosa; often begins on oral mucosa, may lead to hoarseness; skin is usually painful and Nikolsky's sign positive | Light: suprabasilar blister with acantholysis DIF: intercellular IgG and C3 | Corticosteroids (prednisone, 1 mg per kg per day) taper to maintenance; may add azathioprine (Imuran), methotrexate, cyclophosphamide (Cytoxan), mycophenolate mofetil (CellCept); plasmapheresis | May be fatal if untreated, risk of infection in treated and untreated cases |
| Paraneoplastic pemphigus | Very rare, female to male ratio is 2:1, onset at 60 years and older, associated with malignancy, most often lymphoid | Extensive painful mucocutaneous erosions, resembling pemphigus vulgaris, in the presence of neoplasm; targetoid erythematous papules with dusky centers resembling erythema multiforme may also be present | Light: suprabasilar blister with acantholysis (PV-like), with basal cell vacuolation, exocytosis, and dyskeratotic keratinocytes (erythema multiforme-like) | Detection and treatment of the neoplasm | Usually rapidly fatal if associated with a malignant neoplasm |
| DIF: intercellular and basement membrane IgG and C3 | |||||
| IIF: positive for rat bladder (cuboidal epithelium) and stratified squamous epithelium | |||||
| Bullous pemphigoid | Equal in men and women, onset at 60 to 80 years of age, rarely in children | Tense bullae with clear fluid or erosions, may begin as erythematous urticarial, pruritic plaques, localized or generalized on the lower legs, forearms, thighs, groin, abdomen, but rarely on mucosa | Light: subepidermal blister with mixed superficial inflammation DIF: IgG and/or C3 at the basement membrane | Corticosteroids (prednisone) alone or with azathioprine, mycophenolate mofetil or a tetracycline | Self-limited, rarely fatal even if untreated |
| Cicatricial pemphigoid | Rare, female to male ratio is 2:1, onset at 40 to 60 years of age | Bullae rupture within hours leaving painful erosions that heal with scarring; most common sites are oral and conjunctival mucosa; skin involved in one third of cases, head and upper chest | Light: subepidermal blister with mixed infiltrate in fresh lesions and predominant fibroblasts in older, scarring lesions | Corticosteroids; may add dapsone for oral involvement or cyclophosphamide or azathioprine for ocular involvement; topical treatment of oral lesions with steroid gel or dexamethasone mouthwash (Roxane) | Ocular lesions may lead to blindness; oral complications include laryngeal or esophageal strictures with dysphagia, hoarseness, loss of voice, or need for tracheostomy |
| DIF: linear IgG and C3 on the basement membrane, +/− IgA or IgM treatment of oral lesions | |||||
| Dermatitis herpetiformis (dapsone) | Rare, male to female ratio is 2:1, onset at 20 to 40 years of age, but also in children, whites, rare in blacks or Asians | Grouped (herpetiform) excoriations or vesicles symmetrically located on extensor remission; watch for signs surfaces of elbows, knees, sacrum, buttocks, and shoulders with intense pruritus and burning sensation | Light: neutrophilic abscesses in dermal papillae, dermal infiltrates of neutrophils and eosinophils with subepidermal vesicles | Gluten-free diet; sulfones dermal infiltrates of neutrophils and eosinophils | Chronic with occasional remission; watch for signs of other autoimmune disorders; may coexist with gluten-sensitive enteropathy |
| DIF: granular IgA deposits in the tips of the dermal papillae | |||||
| Linear IgA dermatosis (LAD) or chronic bullous disease of childhood (CBDC) | LAD: equal in men and women, onset after puberty, usually in 30s | LAD: pruritic symmetric grouped annular crusted papules, vesicles, or bullae on extensor surfaces, elbows, knees, or buttocks | Light : subepidermal bullae with infiltrate of neutrophils at basement membrane and dermal papillae tips | Sulfones (dapsone) +/− low-dose prednisone | LAD: variable course |
| CBDC: onset before age five | CBDC: rings of grouped bullae around old lesions on genitalia, face, or perioral area | DIF: linear deposits of IgA along the basement membrane, +/− IgG and C3 | CBDC: usually clears in two years or less |
Light = light microscopy; DIF = direct immunofluorescence; IIF = indirect immunofluorescence; LAD = linear IgA dermatosis; CBDC = chronic bullous disease of childhood.
Pemphigus Vulgaris
Pemphigus encompasses a group of auto-immune blistering diseases of the skin and mucous membranes. Included in this group is pemphigus vulgaris, a bullous disease involving the skin and mucous membranes, which may be fatal if not treated with appropriate immunosuppressive agents. The detection of circulating antibodies against keratinocyte cell surfaces led to the understanding that pemphigus was an autoimmune disease.1
According to several retrospective studies,2 the prevalence of pemphigus vulgaris is equal in men and women. Although it may be seen in children and the elderly, the mean age of onset is between 40 and 60 years. Pemphigus vulgaris is also more common in persons of Jewish and Mediterranean descent.
Characteristically, lesions start in the oral mucosa, followed by the appearance of skin lesions months later. The bullae on the skin may remain localized for six to 12 months, then subsequently become widespread. Rarely, the lesions may arise as a generalized acute eruption. The lesions can be pruritic but are usually painful and accompanied by a burning sensation. Mouth lesions may be tender, preventing adequate food intake that leads to weight loss. The lesions may be accompanied by weakness and malaise, and a history of epistaxis, dysphagia, and hoarseness.
The primary lesion on the skin is a flaccid blister. These blisters are fragile, rupture easily and, therefore, are not often seen. More likely to be noticed are the painful erosions that are the result of broken blisters (Figure 1). These erosions bleed easily and often become crusted. The lesions are round to oval in shape, and range from skin-colored to erythematous. Nikolsky's sign, in which the epidermis is easily detached from the skin, is elicited by applying lateral pressure to a bulla, leading to lateral extension of the blister, and is usually positive. Sites of predilection include the scalp, face, chest, axillae, groin, and umbilicus.
FIGURE 1.
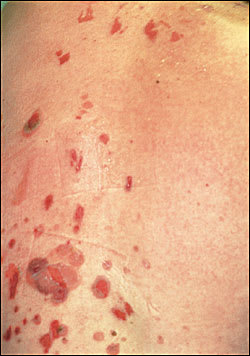
Pemphigus vulgaris. Erosions and flaccid bullae on normal skin.
Painful erosions, most often in the oral cavity, are seen in nearly all patients with pemphigus vulgaris (Figure 2). The buccal mucosa is the most common site of involvement in the oral cavity. Other sites of mucous membrane involvement include the pharynx and larynx, which is manifested by hoarseness. The conjunctiva, esophagus, anus, penis, vagina, and labia have also been reported as sites where painful erosions are located.
FIGURE 2.
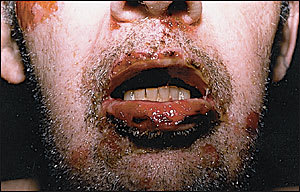
Pemphigus vulgaris. Involvement of the oral mucosal membranes.
Biopsy of the margin of a bulla, when examined by light microscopy, will reveal a suprabasilar blister with acantholysis. The acantholysis is caused by a loss of cohesion between cells in the lower epidermis, resulting in the formation of a blister just above the basal cell layer. Early pemphigus vulgaris lesions may show eosinophilic spongiosis as well.
Intercellular deposits of IgG and C3 are the defining signs of pemphigus vulgaris. Thus, there is intercellular staining throughout the epidermis on direct immunofluorescence.
TREATMENT
Corticosteroids are the mainstay of treatment for patients with pemphigus vulgaris. Prednisone (1 mg per kg per day), with or without other immunosuppressive agents, should be initiated immediately. It should be continued until there is cessation of new bullae formation and Nikolsky's sign can no longer be elicited. The dosage is then reduced by one half until all of the lesions have cleared, followed by tapering to a minimum effective maintenance dosage.
Other immunosuppressive agents used in conjunction with corticosteroids include azathioprine (Imuran), methotrexate, cyclophosphamide (Cytoxan), and mycophenolate mofetil (CellCept). Because it may take several weeks for the immunosuppressive agents to work, some physicians start these agents concurrently with prednisone. In severe cases, plasmapheresis may be required.
COURSE AND COMPLICATIONS
Even with the use of corticosteroids and other immunosuppressive agents, there is still significant morbidity and mortality associated with pemphigus vulgaris. A common cause of death is infection secondary to the immunosuppression required to treat the disease. Most deaths occur within the first few years of the disease.2 Unfortunately, many of the drugs used to treat this disease have serious side effects, and patients must be monitored closely for infection, renal and liver function abnormalities, electrolyte disturbances, hypertension, diabetes, anemia, and gastrointestinal bleeding.
Paraneoplastic Pemphigus
Paraneoplastic pemphigus is an extremely rare entity that has an onset at 60 years or older and is more common in women than men. It is distinct from the classic forms of pemphigus and is characterized by extensive mucocutaneous erosions in the presence of a neoplasm, most often a leukemia or a lymphoma.3 Other associated neoplasms, malignant and benign, include Waldenström's macroglobulinemia, sarcomas, thymomas, and Castleman's disease.4
The predominant feature of paraneoplastic pemphigus is painful mucous membrane erosions, of which oral erosions are the first sign of disease in 22.2 percent of cases.5 The most common sites involved are the lips and oral mucosa, with multiple, severe, persistent erosions. Symptoms of oropharyngeal involvement may include sore throat and dysphagia. Bilateral conjunctival involvement has been noted in up to 72.2 percent of cases.5 The skin lesions vary in shape and size, with a confluent erythema of the trunk, on which blisters and erosions form. Erythematous maculopapular lesions with dusky centers or central vesicles may arise on the extremities, mimicking target lesions seen in erythema multiforme. Occasionally, the lesions may be pruritic.
On histopathologic examination, paraneoplastic pemphigus appears to be a combination of pemphigus vulgaris and erythema multiforme. There is suprabasilar acantholysis as seen in pemphigus vulgaris, as well as basal cell vacuolation, lymphocytic exocytosis, and dyskeratotic keratinocytes typical of erythema multiforme.
Paraneoplastic pemphigus is distinguished from the other forms of pemphigus as direct immunofluorescence reveals not only IgG and C3 deposits within the intercellular spaces but also along the basement membrane zone.
In the classic forms of pemphigus, indirect immunofluorescence is positive only on stratified squamous epithelial substrates. However, in paraneoplastic pemphigus, there is staining of other tissues, including the bladder, heart, and liver. IgG autoantibodies are directed against desmoplakins I and II (components of the cytoplasmic plaque), which are present in stratified squamous epithelium and these other tissues.
TREATMENT
There is little to offer in the treatment of paraneoplastic pemphigus. If a benign tumor is resected, some patients may go into remission. Unfortunately, the prognosis is generally poor, and treatment is usually unsuccessful. Immunosuppressive treatment and plasmapheresis have not been effective; however, immunophoresis may be a promising alternative.6
COURSE AND COMPLICATIONS
Paraneoplastic pemphigus is a rapidly progressive bullous disease that is invariably fatal when associated with a malignant tumor. When paraneoplastic pemphigus occurs in the context of a benign neoplasm, the mucocutaneous erosions will usually show gradual resolution after excision of the tumor. It is important to remember that paraneoplastic pemphigus may precede the clinical appearance of a neoplasm; therefore, it is mandatory that these patients receive screening for neoplasms and regular follow-up care.
Bullous Pemphigoid
Bullous pemphigoid is an autoimmune skin disorder characterized by subepidermal blistering that results in large, tense bullae. It occurs mainly in the elderly and rarely in children. Onset is typically between 60 and 80 years of age. There is equal incidence in men and women, and there are no known racial or ethnic predilections. In France and Germany, the incidence is estimated at seven per 1 million per year.7
The lesions of bullous pemphigoid may initially start as an urticarial eruption (Figure 3), which over a course of weeks to months, develops into bullae. The lesions are usually pruritic, and there may be tenderness at the site of eroded lesions.
FIGURE 3.
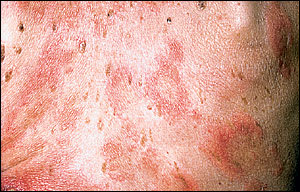
Bullous pemphigoid. Urticarial plaques.
Once formed, blisters are large and tense, with a round or oval shape. Discrete lesions arise on normal or erythematous skin (Figure 4) and are scattered throughout the body, including the axillae, medial thighs, groin, abdomen, flexor forearms, and lower legs. The lesions may be localized or generalized. Most often, the bullae are filled with a clear fluid, but they also can be hemorrhagic. There is no scar formation noted following the lesions of bullous pemphigoid, but milia may appear at sites of previously involved skin.
FIGURE 4.
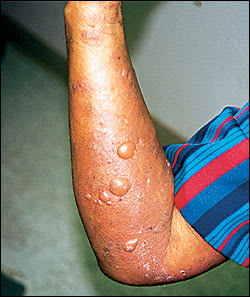
Bullous pemphigoid. Tense bullae on erythematous skin.
The involvement of mucous membranes is much less common with bullous pemphigoid than in pemphigus vulgaris, with blisters that are less easily ruptured. Sites involved include the oral cavity, anus, and genital mucosa.
Histologic examination of a skin biopsy from a bulla reveals a subepidermal blister with superficial dermal inflammation consisting of lymphocytes, histiocytes, and eosinophils. Urticarial lesions may also be accompanied by papillary dermal edema.
On electron microscopy, blister formation is found to occur within the lamina lucida of the basement membrane, causing a loss of anchoring filaments and hemidesmosomes. Direct immunofluorescence reveals deposition of IgG, and possibly C3, along the basement membrane zone in a linear pattern.
TREATMENT
Treatment consists of systemic prednisone, alone or in combination with a steroid-sparing agent such as azathioprine, mycophenolate mofetil or a tetracycline. These drugs are usually started simultaneously, followed by a gradual tapering of the prednisone and continuation of the steroid-sparing agent until clinical remission is achieved. Mild cases may require only topical corticosteroids. Methotrexate may be used in patients with severe disease who are unable to tolerate prednisone.
COURSE AND COMPLICATIONS
Bullous pemphigus is a self-limited disease, but may last from months to years. It is rarely fatal, and even without corticosteroid therapy, carries a good prognosis. Approximately one half of treated cases will remit within six years.7
Cicatricial Pemphigoid
Cicatricial pemphigoid is a rare, blistering disease of the skin, characterized by severe, erosive lesions of the skin and mucous membranes. Mucous membrane involvement is common, primarily of the oral mucosa and conjunctiva, but may also include the nasopharynx, larynx, esophagus, genitalia, and rectal mucosa. Skin involvement occurs in one third of patients and is focused around the scalp, face, and upper trunk, and heals with scars. The bullae are tense, and located on an erythematous or urticarial base (Figure 5). The vesiculobullous lesions tend to rupture within hours, leaving painful erosions and ulcers that can easily become secondarily infected. Ocular cicatricial pemphigoid is characterized by chronic conjunctivitis that leads to decreased vision, photosensitivity, and scarring and fibrosis that can eventually cause blindness (Figure 6).
FIGURE 5.
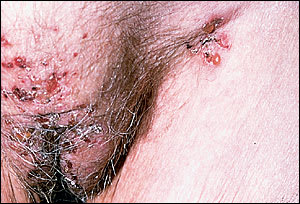
Cicatricial pemphigoid. Tense bullae on erythematous skin.
FIGURE 6.
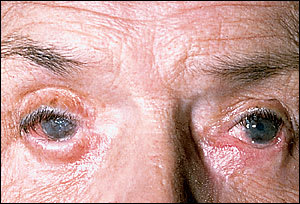
Cicatricial pemphigoid. Symble-pharon formation.
There is a 2:1 preponderance for women, and the age of onset is typically in late adulthood, most often between 40 and 60 years of age.7
Lesions of the oral mucosa may be seen on the gingiva, buccal mucosa, palate, tongue, and lips. The lesions consist of tense blisters that rupture easily and erosions. In severe cases of cicatricial pemphigoid, there may be adhesions between the various structures of the oral cavity involved, and gingival involvement can cause dental complications.
Other mucous membranes that can be affected include those of the nasopharynx, larynx, and esophagus. Laryngeal involvement may lead to a sore throat, hoarseness, and possible loss of speech. Supraglottic stenosis secondary to erosions, scarring, and edema may necessitate a tracheostomy as the airway is further compromised. Esophageal erosions and scarring, which occurs in 8 percent of cases,8 can lead to the formation of strictures, and these patients may present with dysphagia, odynophagia, and weight loss. Eventually, complete occlusion of the esophagus may occur.
The blisters are subepidermal and surrounded by a mixed inflammatory cell infiltrate. In mucosal lesions, this infiltrate is primarily made up of mononuclear cells, histiocytes and plasma cells, while the skin lesion infiltrate is composed predominantly of eosinophils and neutrophils. Older skin lesions have less inflammation, with prominent fibroblast proliferation.
Direct immunofluorescence reveals linear deposition of C3 and IgG continuously along the basement membrane. IgA and IgM may also be detected. These findings are observed in unaffected and perilesional skin.
TREATMENT
Treatment of mild lesions of the skin and oral mucosa consists of topical corticosteroids in a gel or occlusive base, which is best used before bedtime. A swish and spit dexamethasone (Roxane) mouthwash can be helpful for oral lesions. Dapsone has been shown to be of benefit in some cases. In severe cases of cicatricial pemphigoid, systemic steroids are prescribed, with or without dapsone. Because of the severity of the sequelae of these lesions, aggressive early treatment is essential. For severe ocular involvement, patients are treated with systemic steroids, along with cyclophosphamide or azathioprine. Prednisone is usually given for three to six months, with the cyclophosphamide or azathioprine continued for one year. At that point, the medications may be decreased, and if the patient remains free of disease, the medications may be discontinued.
COURSE AND COMPLICATIONS
Cicatricial pemphigoid is a chronic progressive disease that rarely remits spontaneously. Early immunosuppressive treatment is necessary. All patients will need thorough ophthalmologic and dermatologic examinations, and consultations with otolaryngology, gastroenterology, and gynecology specialists may also be appropriate.
Dermatitis Herpetiformis
Dermatitis herpetiformis is an intensely pruritic, chronic skin disease characterized by papulovesicular lesions and urticarial wheals located on the extensor surfaces in a symmetric distribution. The disease persists indefinitely, and is associated with a gluten-sensitive enteropathy in most patients.
The incidence of dermatitis herpetiformis is 10 to 39 cases per 100,000 persons. Onset tends to be between 20 and 40 years of age but may occur at any age, including childhood, and there is a 2:1 preponderance for men.7 Dermatitis herpetiformis is principally a disease that affects whites. It rarely occurs in blacks or Asians.
The lesions of dermatitis herpetiformis usually begin as a vesicle, but may also be erythematous papules, urticarial-like wheals, excoriations, crusts, or rarely, large bullae. The lesions may be grouped, giving a “herpetiform” appearance. Once the lesions have resolved, there may be transient hyper- or hypopigmentation. The lesions are usually intensely pruritic, accompanied by burning and stinging. Many patients experience localized burning, stinging, and pruritus approximately eight to 12 hours before the onset of lesions, and many are able to predict an eruption. Rarely, the lesions may be asymptomatic. There is symmetric distribution along the extensor surfaces, including the elbows, knees, buttocks, shoulders, and sacral areas. Less frequently, the lesions are found on the scalp, face, hairline, and the posterior neck. Involvement of the palms and soles is rare, and mucous membrane lesions are uncommon.
Dermatitis herpetiformis is characterized histopathologically by neutrophilic microabscesses in dermal papillae, dermal infiltration of neutrophils and eosinophils, and the formation of subepidermal vesicles. Blisters form within the lamina lucida. Dermal blood vessels may be surrounded by a lymphohistiocytic infiltrate as well.
Direct immunofluorescence reveals granular deposition of IgA in the tips of dermal papillae. In areas corresponding to IgA deposits, there may also be complement deposition. IgA and IgG antireticulin and antien-domysial antibodies have been detected in dermatitis herpetiformis patients' sera.9 An increased incidence of antinuclear and antithyroid microsomal antibodies are also found in these patients.
TREATMENT
Patients will experience prompt relief of lesions within one to two days of initializing treatment with dapsone or sulfapyridine.10 It is important to remember to always check glucose-6-phosphate dehydrogenase (G6PD) and baseline complete blood count levels before starting dapsone, followed by complete blood cell counts every month to monitor for signs of hemolytic anemia. A slight decrease in hemoglobin is common.
Other methods of treatment include dietary modification. One form is the gluten-free diet, which has been found to improve both intestinal and skin lesions. The onset is slow, taking from five months to one year before the effect is noted; however, close adherence to the diet will allow patients to significantly decrease or stop using the medications. Another diet that has been found to alleviate skin lesions is the elemental diet, consisting of free amino acids, short chain polysaccharides and small amounts of triglycerides. Alleviation of skin lesions can occur within a few weeks of starting the diet, even if the patient ingests large amounts of gluten, but this diet is difficult to tolerate.
COURSE AND COMPLICATIONS
Dermatitis herpetiformis follows a prolonged course, lasting up to years. Approximately one third of patients eventually have spontaneous remission.9 Dermatitis herpetiforms responds well to medications and diet, and has a good prognosis. Associated manifestations that may cause complications include the gluten-sensitive enteropathy, which can cause steatorrhea, abnormald-xylose absorption, and anemia. There has also been an increased incidence of atrophic gastritis and achlorhydria in patients with gluten sensitivity. Reports of increased frequency of gastrointestinal lymphomas have also been noted, from which the gluten-free diet has been found to be protective. Patients with dermatitis herpetiformis have also been found to have an increased incidence of other autoimmune disorders, including thyroid disease, type 1 diabetes mellitus, systemic lupus erythematosus, vitiligo, and Sjögren's syndrome.
Linear IgA Disease
Linear IgA dermatosis is a rare autoimmune bullous disorder, characterized by linear deposition of IgA along the basement membrane.11 It was originally thought to be a manifestation of dermatitis herpetiformis; however, based on immunopathology and immunogenetics, it is now known that linear IgA dermatosis is a distinct entity. Chronic bullous disease of childhood shares the same linear deposition of IgA along the basement membrane and is believed to be a variant of linear IgA dermatosis.
Linear IgA dermatosis most commonly presents in patients older than 30 years.11 Chronic bullous disease of childhood occurs in young children, usually presenting in those younger than five years.12
The lesions of linear IgA dermatosis consist of pruritic, annular papules, vesicles, and bullae that are found in groups. There is a predilection for the extensor surfaces, with symmetric distribution. Lesions are seen on the elbows, knees, and buttocks. Because of itching, excoriations will lead to the formation of many crusted papules.
Chronic bullous disease of childhood presents with abrupt onset of tense bullae on an inflamed, erythematous base and is accompanied by pruritus and a burning sensation. The lesions are most frequently found on or near the genitalia, but may also be found on other areas, including the face, especially the perioral region. Oral ulcers are noted in 50 percent of cases.12 Characteristic “collarettes” of blisters often form as new lesions arise in the periphery of old lesions.
In both forms of linear IgA dermatosis, mucous membrane involvement may occur and ranges in severity from mild oral ulcers to severe oral or conjunctival disease.
On histopathology, in linear IgA dermatosis and chronic bullous disease of childhood, the bullae are subepidermal, with collections of neutrophils along the basement membrane and occasionally in the dermal papillary tips. On direct immunofluorescence, deposition of IgA in a linear pattern is noted along the basement membrane. There may also be deposition of IgG and C3.
TREATMENT
Skin lesions in linear IgA dermatosis and chronic bullous disease of childhood respond rapidly when treated with dapsone or sulfa-pyridine. Again, there is a risk of hemolytic anemia in G6PD-deficient patients. Some patients may require low-dose prednisone initially to suppress blister formation.
COURSE AND COMPLICATIONS
The course of linear IgA dermatosis is variable and unpredictable. The disease may spontaneously remit in some cases; however, it may last for years with few episodes of remission in others. Chronic bullous disease of childhood follows a much different course, with resolution occurring within two years of onset in most cases.
