In approximately 5 percent of sudden cardiac deaths, no demonstrable anatomic abnormality is found. Some cases are caused by sudden arrhythmia death syndrome. A prolonged QT interval is a common thread among the various entities associated with sudden arrhythmia death syndrome. A number of drugs are known to cause QT prolongation (e.g., terfenadine), as are hypokalemia, hypomagnesemia, myocarditis, and endocrine and nutritional disorders. Recently, attention has focused on a group of inherited gene mutations in cardiac ion channels that cause long QT syndrome and carry an increased risk for sudden death. Some of the highest rates of inherited long QT syndrome occur in Southeast Asian and Pacific Rim countries. The median age of persons who die of long QT syndrome is 32 years; men are predominately affected. In addition to a prolonged QT interval, which occurs in some but not all persons with long QT syndrome, another characteristic electrocardiographic abnormality is the so-called Brugada sign (an upward deflection of the terminal portion of the QRS complex). Most cardiac events are precipitated by vigorous exercise or emotional stress, but they also can occur during sleep. Torsades de pointes and ventricular fibrillation are the usual fatal arrhythmias. Long QT syndrome should be suspected in patients with recurrent syncope during exertion and those with family histories of sudden, unexpected death. Unfortunately, not all persons with long QT syndrome have premonitory symptoms or identifiable electrocardiographic abnormalities, and they may first present with sudden death. Beta blockers, potassium supplements, and implantable defibrillators have been used for treatment of long QT syndrome. Identifying the specific gene mutation in a given patient with long QT syndrome can help guide prophylactic therapy.
Sudden death from cardiac arrest occurs in approximately 300,000 persons per year in the United States. Coronary artery disease accounts for the majority of cases, while most of the others are associated with anatomic abnormalities and are presaged by clinical signs and symptoms (Table 1). Occasionally, the underlying heart disease remains undiagnosed, and death comes suddenly and unexpectedly, but the postmortem examination reveals a cause. In 1 to 5 percent of deaths, however, no anatomic abnormality can be found, and this group constitutes the newly described “sudden arrhythmia death syndrome” (SADS).1,2
TABLE 1 Causes of Sudden Cardiac Death
| More common |
| Coronary atherosclerosis |
| Cardiomyopathies |
| Aortic valvular stenosis |
| Drug-induced electrolyte imbalance |
| Viral myocarditis |
| Less common |
| Sudden arrhythmia death syndrome |
| Anomalous coronary artery origin |
| Mitral ball-valve thrombus or myxoma |
| Tumors of atrioventricular node region |
| Hepatic steatosis-induced arrhythmia |
| Wolff-Parkinson-White syndrome |
| High-degree atrioventricular heart block (Stokes-Adams syndrome) |
| Sick sinus syndrome |
| Idiopathic ventricular fibrillation |
SADS encompasses a number of clinical entities, including congenital long QT syndrome, Wolff-Parkinson-White syndrome, idiopathic ventricular fibrillation, and coronary artery spasm (e.g., from cocaine intoxi cation). Some cases of sudden infant death syndrome also have been attributed to fatal arrhythmias during sleep. Arrhythmias may be induced in normal hearts by drugs (e.g., terfenadine [Seldane]); electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia); myocarditis; and endocrine, central nervous system, or nutritional disorders. These arrhythmias are associated with prolongation of the electrocardiographic QT interval.3 Another class of arrhythmias that is often associated with long QT syndrome includes hereditary genetic defects that affect the cardiac ion channels; this article focuses on this emerging entity.
Illustrative Case
During the night, the screams of a 57-year-old Taiwanese woman awakened her husband, who found her in bed cyanotic, unresponsive, and pulseless. He called for help and administered cardiopulmonary resuscitation (CPR). On arrival, emergency medical personnel found the patient to be in ventricular fibrillation. They administered defibrillatory shocks, and after a period of asystole, the patient's heart returned to sinus rhythm.
The patient's medical history was significant for several episodes of syncope. A previous work-up had discovered frequent premature ventricular complexes and runs of bigeminy on a Holter monitor examination. Quinidine and a beta-adrenergic blocker were prescribed, but at some point the patient had stopped taking them.
On arrival in the hospital emergency department, examination revealed signs of decerebration with dilation of both pupils. Electrocardiography (ECG) showed no abnormality, with normal PR and QT intervals. Slight elevations of serum troponin I and creatine kinase MB levels were attributed to prolonged CPR. A second cardiac arrest that began with asystole was followed by ventricular fibrillation. Junctional bradycardia occurred several hours after a second cardiac resuscitation, followed by electromechanical dissociation, lactic acidosis, and death 11 hours after the initial event.
Autopsy demonstrated a heart of normal weight with no anatomic abnormalities. Coronary arteries were examined at 0.5-cm intervals and were found to be without significant luminal narrowing. Examination of other organs, including the brain, showed only old scarring of the middle lobe of the right lung.
Historical Background of SADS
A sudden, unexpected nocturnal death syndrome has been known in Southeast Asian and Pacific Rim countries for decades. The fatal event typically is accompanied by labored respiration, gasping, or groaning. It is called “bangungut” in the Philippines,“pokkuri” in Japan, and “lai tai” in Thailand, all meaning “sleep death.” In these areas it is recognized as a leading cause of death in young men, with the highest mortality rate occurring in northeastern Thailand (40 per 100,000 persons per year).4 The degree of familial expression can be devastating. One report5 documented 25 sudden deaths in one family, with 16 of them occurring during the night. A U.S. study4 of 82 instances of SADS among refugees from Southeast Asia showed an age range of 16 to 63 years, with a median age of 32 years. All but one of the victims were men.
Discovery of a characteristic hereditary ECG abnormality, the Brugada sign, in right precordial leads resulted in classification of this condition as a right ventricular repolarization defect.6,7 The Brugada sign consists of a prominent upward deflection at the end of the QRS complex, which may be associated with right bundle branch block (Figure 1).7 Hereditary prolongation of the QT interval (Figure 2) also has been associated with SADS.
FIGURE 2.
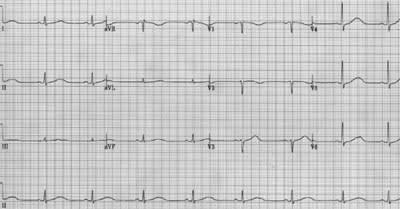
Long QT interval shown in all electrocardiographic leads. The interval is measured from the beginning of the QRS complex to the end of the T wave. In this patient, the QT interval is 560 msec. The normal upper limit for the QT interval is 440 msec in men and 460 msec in women. This electrocardiogram compares with Figure 1 showing a normal QT interval.
The most common type of SADS associated with hereditary long QT syndrome is the autosomally dominant Romano-Ward syndrome (syncope, seizures, and sudden death).8,9 While the syndrome may first manifest as syncope or seizures, sudden death is the only event in 30 to 40 percent of patients.6 A number of known genetic errors are associated with this syndrome. The affected genes make proteins that are involved in the function of cardiac cell membrane sodium and potassium ion channels.
ECG Manifestations of Inherited Long QT Syndrome
When ECGs could be obtained during SADS events, they have always shown the polymorphic ventricular tachyarrhythmia termed “torsades de pointes” (Figure 3), or ventricular fibrillation.4 More important from the standpoint of prophylactic intervention are findings that may be present in asymptomatic persons before a terminal event occurs. QT prolongation is characteristic, with a QTc greater than 440 msec in men and greater than 460 msec in women (Figure 2). However, in some cases a borderline prolonged QT interval is seen. Another indicative finding is T-wave alternans (alternating polarity and amplitude), which may be present at rest for brief moments but most commonly appears during emotional or physical stress. The severity of T-wave alternans tends to correlate with the risk of cardiac events.10
FIGURE 3.
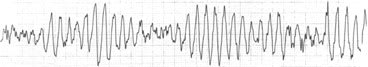
Electrocardiogram showing torsades de pointes. Note the more or less symmetric variation in the ventricular tachycardia waves. Torsades de pointes may resolve into sinus rhythm or progress into ventricular fibrillation.
Approximately 60 percent of persons with familial long QT syndrome are symptomatic.11 In these persons, sinus pauses, poor response of heart rate to exercise, and syncope may occur. Alterations in different ion channels lead to abnormalities in the refractory period and repolarization of cardiac pacemaker cells, promoting ventricular tachyarrhythmias.
When present, the long QT syndrome permits recognition of asymptomatic carriers of the long QT syndrome gene. However, not all gene carriers who are at risk of cardiac events have abnormal findings on ECG.11,12
To date, five different gene mutations have been identified in patients with inherited long QT syndrome. Four genes form potassium channels, and one forms the cardiac sodium channel.2,11 Mutations result in defective proteins that form an abnormal ion channel, thereby reducing current flow through the cell membrane and lengthening the QT interval.13
Clinical Screening for Long QT Syndrome
In long QT syndrome, most life-threatening cardiac events are associated with physical or emotional stress, but deaths also can occur during sleep, and sleep-related deaths tend to cluster in families. Review of the International LQTS (long QT syndrome) Registry, which was established in 1979, has shown that acute arousal events (e.g., vigorous exercise, emotional stress, loud noise) often occurred just before a SADS episode. Slowing of the heart rate during sleep leads to a prolonged QT interval in some affected persons, which may explain the nocturnal deaths that occur among persons with SADS.14
Clues to the diagnosis of SADS are listed in Table 2. In particular, this syndrome should be suspected in any patient who has recurrent syncopal or near-syncopal spells during physical activity or emotional distress, or who has a family history of unexplained sudden death in an otherwise-healthy young person or infant.
TABLE 2 Clues to Sudden Arrhythmia Death Syndrome
| Otherwise healthy person with: | |
| Syncope, especially with exercise or severe stress | |
| Family history of sudden, unexpected death (nocturnal, swimming, athletic exertion, startle response, SIDS) | |
| Pacific Rim origin (Southeast Asia, Japan, Philippines) | |
| ECG: long QT syndrome not otherwise explained, Brugada sign, torsades de pointes, ventricular fibrillation | |
SIDS = sudden infant death syndrome; ECG = electrocardiography.
In some persons, resting ECG can detect long QT syndrome. However, prolongation of the QT interval has been minimal or absent in a substantial number of persons known to carry genetic mutations that cause long QT syndrome. If the resting ECG is nondiagnostic, ECG should be performed during exercise. QT intervals that are borderline or normal at rest may become distinctly abnormal with exercise.
Risk Assessment
The most common mutations related to the long QT syndrome are in three genetic loci termed LQT1 and LQT2 (potassium channel genes) and LQT3 (a sodium channel gene). Risk stratification for these patients is based on the locus, length of the QTc interval with a cutoff of 500 msec, and gender. In one large study,15 mutations of LQT1 were the least hazardous, with a 30 percent probability of cardiac events by age 40 years. The QTc is QT corrected for heart rate, and cardiac event was defined as syncope, cardiac arrest, or sudden death. LQT2 and LQT3 mutations were associated with event rates approaching 50 percent. QTc of more than 500 msec is a risk marker for LQT1 for either gender. Men with LQT2 mutations and QTc of less than 500 msec have relatively low risk of cardiac events, whereas all women with LQT2 mutations have relatively high risk. All patients with mutations at the LQT3 locus have relatively high risk of cardiac events. Prophylactic beta-blocker therapy has been recommended for high-risk patients, and decisions may be individualized for low-risk patients.15
Treatment
Competitive sports carry some inherent risk in patients with long QT syndrome, because symptoms often are precipitated by physical exertion or emotion. Given the rare nature of SADS, it is understandable that drug treatment recommendations are mostly anecdotal. The goal of therapy is the resolution of symptoms and, in many cases, abnormalities on ECG normalize after treatment. Different ion channel gene mutations are targeted with different medications. Beta blockers and potassium supplements are among the drugs used to treat long QT syndrome. Permanent pacemakers or automatic implantable defibrillators also have been employed.
Measures to prevent long QT syndrome include avoiding hypokalemia and medications that lengthen the QT interval.
The future holds promise of “mutation-specific pharmacology.”14,16 Thus, identification of the type of mutation could be useful in determining optimal therapy and may become more generally applicable when analysis of the long QT syndrome gene moves from the research laboratory into community medicine.
In conclusion, hereditary ion channel disorders are now recognized as a cause of SADS. These conditions, comprising long QT syndrome and the Brugada syndrome, account for a substantial proportion of sudden cardiac deaths when no anatomic abnormalities are found. Primary care physicians have an opportunity to recognize the occasional patient with a family history or premonitory signs and symptoms of SADS, allowing for the initiation of appropriate intervention to prevent a potentially fatal event.