Hemolysis presents as acute or chronic anemia, reticulocytosis, or jaundice. The diagnosis is established by reticulocytosis, increased unconjugated bilirubin and lactate dehydrogenase, decreased haptoglobin, and peripheral blood smear findings. Premature destruction of erythrocytes occurs intravascularly or extravascularly. The etiologies of hemolysis often are categorized as acquired or hereditary. Common acquired causes of hemolytic anemia are autoimmunity, microangiopathy, and infection. Immune-mediated hemolysis, caused by antierythrocyte antibodies, can be secondary to malignancies, autoimmune disorders, drugs, and transfusion reactions. Microangiopathic hemolytic anemia occurs when the red cell membrane is damaged in circulation, leading to intravascular hemolysis and the appearance of schistocytes. Infectious agents such as malaria and babesiosis invade red blood cells. Disorders of red blood cell enzymes, membranes, and hemoglobin cause hereditary hemolytic anemias. Glucose-6-phosphate dehydrogenase deficiency leads to hemolysis in the presence of oxidative stress. Hereditary spherocytosis is characterized by spherocytes, a family history, and a negative direct antiglobulin test. Sickle cell anemia and thalassemia are hemoglobinopathies characterized by chronic hemolysis.
Hemolysis is the destruction or removal of red blood cells from the circulation before their normal life span of 120 days. While hemolysis can be a lifelong asymptomatic condition, it most often presents as anemia when erythrocytosis cannot match the pace of red cell destruction. Hemolysis also can manifest as jaundice, cholelithiasis, or isolated reticulocytosis.
Pathophysiology
There are two mechanisms of hemolysis. Intravascular hemolysis is the destruction of red blood cells in the circulation with the release of cell contents into the plasma. Mechanical trauma from a damaged endothelium, complement fixation and activation on the cell surface, and infectious agents may cause direct membrane degradation and cell destruction.
The more common extravascular hemolysis is the removal and destruction of red blood cells with membrane alterations by the macrophages of the spleen and liver. Circulating blood is filtered continuously through thin-walled splenic cords into the splenic sinusoids (with fenestrated basement membranes), a spongelike labyrinth of macrophages with long dendritic processes.1 A normal 8-micron red blood cell can deform itself and pass through the 3-micron openings in the splenic cords. Red blood cells with structural alterations of the membrane surface (including antibodies) are unable to traverse this network and are phagocytosed and destroyed by macrophages.
History and Physical Examination
Anemia most often is discovered through laboratory tests, but the history and physical examination can provide important clues about the presence of hemolysis and its underlying cause. The patient may complain of dyspnea or fatigue (caused by anemia). Dark urine and, occasionally, back pain may be reported by patients with intravascular hemolysis. The skin may appear jaundiced or pale. A resting tachycardia with a flow murmur may be present if the anemia is pronounced. Lymphadenopathy or hepatosplenomegaly suggest an underlying lymphoproliferative disorder or malignancy; alternatively, an enlarged spleen may reflect hypersplenism causing hemolysis. Leg ulcers occur in some chronic hemolytic states, such as sickle cell anemia.
Diagnostic Testing
HEMATOLOGIC TESTS
Along with anemia, a characteristic laboratory feature of hemolysis is reticulocytosis, the normal response of the bone marrow to the peripheral loss of red blood cells. In the absence of concomitant bone marrow disease, a brisk reticulocytosis should be observed within three to five days after a decline in hemoglobin. In a minority of patients, the bone marrow is able to chronically compensate, leading to a normal and stable hemoglobin concentration. The anemia of hemolysis usually is normocytic, although a marked reticulocytosis can lead to an elevated measurement of mean corpuscular volume, because the average mean corpuscular volume of a reticulocyte is 150 fL.2
Review of the peripheral blood smear is a critical step in the evaluation of any anemia. Along with an assessment for pathognomonic red blood cell morphologies, such as spherocytes or schistocytes, examination of the white blood cells and platelets for coexisting hematologic or malignant disorders is essential.
CHEMISTRY TESTS
The destruction of red blood cells is characterized by increased unconjugated bilirubin, increased lactate dehydrogenase, and decreased haptoglobin levels. Lactate dehy drogenase and hemoglobin are released into the circulation when red blood cells are destroyed. Liberated hemoglobin is converted into unconjugated bilirubin in the spleen or may be bound in the plasma by haptoglobin. The hemoglobin-haptoglobin complex is cleared quickly by the liver, leading to low or undetectable haptoglobin levels.3
FIGURE 1. Hemolytic Anemia
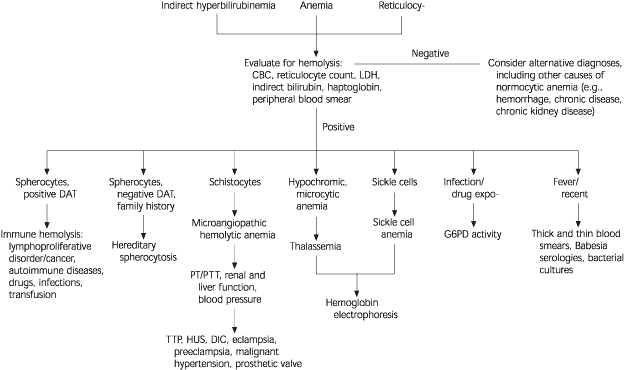
Algorithm for the evaluation of hemolytic anemia. (CBC = complete blood count; LDH = lactate dehydrogenase; DAT = direct antiglobulin test; G6PD = glucose-6-phosphate dehydrogenase; PT/PTT = prothrombin time/partial thromboplastin time; TTP = thrombotic thrombocytopenic purpura; HUS = hemolytic uremic syndrome; DIC = disseminated intravascular coagulation)
TABLE 1 Overview of the Hemolytic Anemias
| Type | Etiology | Associations | Diagnosis | Treatment |
|---|---|---|---|---|
| Acquired* | ||||
| Immune-mediated | Antibodies to red blood cell surface antigens | Idiopathic, malignancy, drugs, autoimmune disorders, infections, transfusions | Spherocytes and positive DAT | Treatment of underlying disorder; removal of offending drug; steroids, splenectomy, IV gamma globulin, plasmapheresis, cytotoxic agents, or danazol (Danocrine); avoidance of cold |
| Microangiopathic | Mechanical disruption of red blood cell in circulation | TTP, HUS, DIC, pre-eclampsia, eclampsia, malignant hypertension, prosthetic valves | Schistocytes | Treatment of underlying disorder |
| Infection | Malaria, babesiosis, Clostridium infections | Cultures, thick and thin blood smears, serologies | Antibiotics | |
| Hereditary† | ||||
| Enzymopathies | G6PD deficiency | Infections, drugs, ingestion of fava beans | Low G6PD activity measurement | Withdrawal of offending drug, treatment of infection |
| Membranopathies | Hereditary spherocytosis | Spherocytes, family history, negative DAT | Splenectomy in some moderate and most severe cases | |
| Hemoglobinopathies | Thalassemia and sickle cell disease | Hemoglobin electrophoresis, genetic studies | Folate, transfusions | |
DAT = direct antiglobulin test; IV = intravenous; TTP = thrombotic thrombocytopenic purpura; HUS = hemolytic uremic syndrome; DIC = disseminated intravascular coagulation; G6PD = glucose-6-phosphate dehydrogenase.
*—Other select causes of acquired hemolysis (not discussed in this article) include splenomegaly, end-stage liver disease/spur cell (acanthocyte) hemolytic anemia, paroxysmal cold hemoglobinuria, paroxysmal nocturnal hemoglobinuria, insect stings, and spider bites.
†—Other select causes of inherited hemolysis (not discussed in this article) include Wilson's disease and less common forms of membranopathy (hereditary elliptocytosis), enzymopathy (pyruvate kinase deficiency), and hemoglobinopathy (unstable hemoglobin variants).
URINARY TESTS
In cases of severe intravascular hemolysis, the binding capacity of haptoglobin is exceeded rapidly, and free hemoglobin is filtered by the glomeruli. The renal tubule cells may absorb the hemoglobin and store the iron as hemosiderin; hemosiderinuria is detected by Prussian blue staining of sloughed tubular cells in the urinary sediment approximately one week after the onset of hemolysis.4 Hemoglobinuria, which causes red-brown urine, is indicated by a positive urine dipstick reaction for heme in the absence of red blood cells.
Acquired Disorders
Once the diagnosis of hemolysis is made on the basis of laboratory and peripheral smear findings (Figure 1), it is necessary to determine the etiology. While most forms of hemolysis are classified as predominantly intravascular or extravascular, the age of onset, accompanying clinical presentation, and co-existing medical problems usually guide the clinician to consider either an acquired or a hereditary cause5,6 (Table 1).
IMMUNE HEMOLYTIC ANEMIA
Immune hemolytic anemias are mediated by antibodies directed against antigens on the red blood cell surface. Microspherocytes on a peripheral smear and a positive direct antiglobulin test are the characteristic findings. Immune hemolytic anemia is classified as autoimmune, alloimmune, or drug-induced, based on the antigen that stimulates antibody- or complement-mediated destruction of red blood cells.
AUTOIMMUNE HEMOLYTIC ANEMIA
Autoimmune hemolytic anemia (AIHA) is mediated by autoantibodies and further subdivided according to their maximal binding temperature. Warm hemolysis refers to IgG autoantibodies, which maximally bind red blood cells at body temperature (37°C [98.6°F]). In cold hemolysis, IgM autoantibodies (cold agglutinins) bind red blood cells at lower temperatures (0° to 4°C [32° to 39.2°F]).
When warm autoantibodies attach to red blood cell surface antigens, these IgG-coated red blood cells are partially ingested by the macrophages of the spleen, leaving microspherocytes, the characteristic cells of AIHA (Figure 2).7 These spherocytes, which have decreased deformability compared with normal red blood cells, are trapped in the splenic sinusoids and removed from circulation.
Cold autoantibodies (IgM) temporarily bind to the red blood cell membrane, activate complement, and deposit complement factor C3 on the cell surface. These C3-coated red blood cells are cleared slowly by the macrophages of the liver (extravascular hemolysis).8 Less frequently, the complete complement cascade is activated on the cell surface, resulting in the insertion of the membrane attack complex (C5b to C9) and intravascular hemolysis.
The direct antiglobulin test (DAT), also known as the direct Coombs' test, demonstrates the presence of antibodies or complement on the surface of red blood cells and is the hallmark of autoimmune hemolysis.9 The patient's red blood cells are mixed with rabbit or mouse antibodies against human IgG or C3. Agglutination of the patient's antibody- or complement-coated red blood cells by anti-IgG or anti-C3 serum constitutes a positive test (Figure 3). Red blood cell agglutination with anti-IgG serum reflects warm AIHA, while a positive anti-C3 DAT occurs in cold AIHA.
Although most cases of autoimmune hemolysis are idiopathic, potential causes should always be sought. Lymphoproliferative disorders (e.g., chronic lymphocytic leukemia, non-Hodgkin's lymphoma) may produce warm or cold autoantibodies. A number of commonly prescribed drugs can induce production of both types of antibodies (Table 2).10 Warm AIHA also is associated with autoimmune diseases (e.g., systemic lupus erythematosus), while cold AIHA may occur following infections, particularly infectious mononucleosis and Mycoplasma pneumoniae infection. Human immunodeficiency virus infection can induce both warm and cold AIHA.11
AIHA should be managed in conjunction with a hematologist. Corticosteroids (and treatment of any underlying disorder) are the mainstay of therapy for patients with warm AIHA. Refractory cases may require splenectomy, intravenous gamma globulin, plasmapheresis, cytotoxic agents, or danazol (Danocrine). All of the aforementioned therapies are generally ineffective for cold AIHA, which is managed most effectively by avoidance of the cold and treatment of any underlying disorder.12 Transfusion therapy in AIHA is challenging, and the most compatible red blood cells (i.e., those with the least cross-reacting antibodies) should be given.9
FIGURE 3.
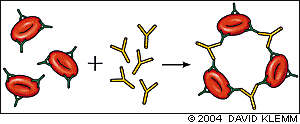
Direct antiglobulin test, demonstrating the presence of autoanti-bodies (shown here) or complement on the surface of the red blood cell. (RBCs = red blood cells)
TABLE 2 Selected Drugs that Cause Immune-Mediated Hemolysis
| Mechanism | Drug absorption (hapten) | Immune complex | Autoantibody |
| DAT Site of hemolysis Medications | Positive anti-IgG Extravascular Penicillin Ampicillin Methicillin Carbenicillin Cephalothin (Keflin)* Cephaloridine (Loridine)* | Positive anti-C3 Intravascular Quinidine Phenacetin Hydrochlorothiazide Rifampin (Rifadin) Sulfonamides Isoniazid Quinine Insulin Tetracycline Melphalan (Alkeran) Acetaminophen Hydralazine (Apresoline) Probenecid Chlorpromazine (Thorazine) Streptomycin Fluorouracil (Adrucil) Sulindac (Clinoril) | Positive anti-IgG Extravascular Alpha-methyldopa Mefenamic acid (Ponstel) Procainamide Ibuprofen Diclofenac (Voltaren) Interferon alfa |
DAT = direct antiglobulin test.
*—Not available in the United States.
Adapted with permission from Schwartz RS, Berkman EM, Silberstein LE. Autoimmune hemolytic anemias. In: Hoffman R, Benz EJ Jr, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, et al., eds. Hematology: basic principles and practice. 3d ed. Philadelphia: Churchill Livingstone, 2000:624.
DRUG-INDUCED HEMOLYTIC ANEMIA
Drug-induced immune hemolysis is classified according to three mechanisms of action: drug-absorption (hapten-induced), immune complex, or autoantibody. These IgG- and IgM-mediated disorders produce a positive DAT and are clinically and serologically indistinct from AIHA.
Hemolysis resulting from high-dose penicillin therapy is an example of the drug-absorption mechanism, in which a medication attached to the red blood membrane stimulates IgG antibody production. When large amounts of drug coat the cell surface, the antibody binds the cell membrane and causes extravascular hemolysis.
Quinine-induced hemolysis is the prototype of the immune complex mechanism, in which the drug induces IgM antibody production. The drug-antibody complex binds to the red blood cell membrane and initiates complement activation, resulting in intravascular hemolysis.
Alpha-methyldopa is the classic example of antierythrocyte antibody induction. Although the exact mechanism is unknown, the drug (perhaps by altering a red blood cell membrane protein and rendering it antigenic13) induces the production of antierythrocyte IgG antibodies and causes an extravascular hemolysis.
ALLOIMMUNE (TRANSFUSION) HEMOLYTIC ANEMIA
The most severe alloimmune hemolysis is an acute transfusion reaction caused by ABO-incompatible red blood cells. For example, transfusion of A red cells into an O recipient (who has circulating anti-A IgM antibodies) leads to complement fixation and a brisk intravascular hemolysis. Within minutes, the patient may develop fever, chills, dyspnea, hypotension, and shock.
Delayed hemolytic transfusion reactions occur three to 10 days after a transfusion and usually are caused by low titer antibodies to minor red blood cell antigens. On exposure to antigenic blood cells, these antibodies are generated rapidly and cause an extravascular hemolysis. Compared with the acute transfusion reaction, the onset and progression are more gradual.14
MICROANGIOPATHIC HEMOLYTIC ANEMIA
Microangiopathic hemolytic anemia (MAHA), or fragmentation hemolysis, is caused by a mechanical disruption of the red blood cell membrane in circulation, leading to intravascular hemolysis and the appearance of schistocytes, the defining peripheral smear finding of MAHA (Figure 4).7
When red blood cells traverse an injured vascular endothelium—with associated fibrin deposition and platelet aggregation—they are damaged and shredded. This fragmentation occurs in a diverse group of disorders, including thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, disseminated intravascular coagulation, preeclampsia, eclampsia, malignant hypertension, and scleroderma renal crisis. In addition, intravascular devices, such as prosthetic cardiac valves and transjugular intrahepatic portosystemic shunts, can induce MAHA.15
INFECTION
Numerous mechanisms link infection and hemolysis.16 Autoantibody induction (e.g., by M. pneumoniae), glucose-6-phosphate dehydrogenase (G6PD) deficiency, and antimicrobial drugs (e.g., penicillin) are discussed elsewhere in this article. In addition, certain infectious agents are directly toxic to red blood cells.
Malaria is the classic example of direct red blood cell parasitization. Plasmodium species, introduced by the Anopheles mosquito, invade red blood cells and initiate a cycle of cell lysis and further parasitization. Both the cellular invasion and the metabolic activity of the parasite alter the cell membrane, leading to splenic sequestration.16,17 Red cell lysis also contributes to the anemia and can be dramatic in the case of “blackwater fever,” named for the brisk intravascular hemolysis and hemoglobinuria that accompany overwhelming Plasmodium falciparum infection. The diagnosis is made by the observation of intracellular asexual forms of the parasite on thick and thin blood smears.
Similarly, Babesia microti and Babesia divergens, tick-borne protozoa, and Bartonella bacilliformis, a gram-negative bacillus transmitted by the sandfly, cause extravascular hemolysis by direct red blood cell invasion and membrane alteration.
Septicemia caused by Clostridium perfringens, which occurs in intra-abdominal infections and septic abortions, causes hemolysis when the bacterium releases alpha toxin, a phospholipase that degrades the red blood cell membrane.
Hereditary Disorders
The mature red blood cell, while biochemically complex, is a relatively simple cell that has extruded its nucleus, organelles, and protein-synthesizing machinery. Defects in any of the remaining components—enzymes, membrane, and hemoglobin—can lead to hemolysis.
ENZYMOPATHIES
The most common enzymopathy causing hemolysis is G6PD deficiency. G6PD is a critical enzyme in the production of glutathione, which defends red cell proteins (particularly hemoglobin) against oxidative damage. This X-linked disorder predominantly affects men. More than 300 G6PD variants exist worldwide, but only a minority cause hemolysis.18
Most patients have no clinical or laboratory evidence of ongoing hemolysis until an event—infection, drug reaction (Table 3),19 or ingestion of fava beans—causes oxidative damage to hemoglobin. The oxidized and denatured hemoglobin cross-links and precipitates intracellularly, forming inclusions that are identified as Heinz bodies on the supravital stain of the peripheral smear. Heinz bodies are removed in the spleen, leaving erythrocytes with a missing section of cytoplasm; these “bite cells” can be seen on the routine blood smear. The altered erythrocytes undergo both intravascular and extravascular destruction. Older red blood cells are most susceptible, because they have an intrinsic G6PD deficiency coupled with the normal age-related decline in G6PD levels.
Hemolysis occurs two to four days following exposure and varies from an asymptomatic decline in hemoglobin to a marked intravascular hemolysis. Even with ongoing exposure, the hemolysis usually is self-limited, as the older G6PD-deficient cells are destroyed. There is no specific therapy other than treatment of the underlying infection and avoidance of implicated medications. In cases of severe hemolysis, which can occur with the Mediterranean-variant enzyme, transfusion may be required.19
G6PD activity levels may be measured as normal during an acute episode, because only nonhemolyzed, younger cells are assayed. If G6PD deficiency is suspected after a normal activity-level measurement, the assay should be repeated in two to three months, when cells of all ages are again present.20
TABLE 3 Agents that Precipitate Hemolysis in Patients with G6PD Deficiency
| Acetanilid* | Phenylhydrazine |
| Furazolidone (Furoxone) | Primaquine |
| Isobutyl nitrite | Sulfacetamide |
| Methylene blue | Sulfamethoxazole (Gantanol) |
| Nalidixic acid (NegGram) | Sulfapyridine |
| Naphthalene | Thiazolesulfone |
| Niridazole* | Toluidine blue |
| Nitrofurantoin (Furadantin, Macrobid, Macrodantin) | Trinitrotoluene (TNT) |
| Phenazopyridine (Pyridium) | Urate oxidase |
*—Not available in the United States.
Adapted with permission from Beutler E. G6PD deficiency. Blood 1994;84:3614.
MEMBRANOPATHIES
Hereditary spherocytosis is an autosomal dominant disorder caused by mutations in the red blood cell membrane skeleton protein genes. With a weakened protein backbone anchoring its lipid bilayer, the membrane undergoes a progressive deterioration in structure, resulting in a spherocyte, the characteristic abnormality seen on peripheral smear. As with AIHA, the spherocytes are unable to pass through the splenic cords and are degraded and ingested by the monocyte-macrophage system.
Although there is marked variability in phenotype, hereditary spherocytosis is typically a chronically compensated, mild to moderate hemolytic anemia. The diagnosis is based on the combination of spherocytosis noted on peripheral smear, a family history (in 75 percent of cases), and a negative DAT. The mean corpuscular hemoglobin concentration frequently is elevated.2,21
Splenectomy effectively arrests the extravascular hemolysis and prevents its long-term complications, such as cholelithiasis and aplastic crises. Because of the inherent risk of infections and sepsis, however, splenectomy generally is reserved for use in patients older than five years with moderate to severe disease, characterized by hemoglobin concentrations of less than 11 g per dL (110 g per L) and jaundice.21–23 Partial splenectomy has been demonstrated to be effective in decreasing hemolysis while maintaining the phagocytic function of the spleen.21,24,25 [Reference 25—strength of recommendation level C, case series]
HEMOGLOBINOPATHIES
Chronic hemolysis can be a characteristic of disorders of hemoglobin synthesis, including sickle cell anemia and thalassemias.
The thalassemias are a heterogeneous group of inherited multifactorial anemias characterized by defects in the synthesis of the alpha or beta subunit of the hemoglobin tetramer (α2β2). The deficiency in one globin chain leads to an overall decrease in hemoglobin and the intracellular precipitation of the excess chain, which damages the membrane and leads to clinically evident hemolysis in the severe forms of alpha thalassemia (hemoglobin H disease) and beta thalassemia (intermedia and major). Beta thalassemia can be diagnosed by hemoglobin electrophoresis, which shows elevated levels of hemoglobins A2 and F, while diagnosis of alpha thalassemia requires genetic studies. Thalassemias are characterized by hypochromia and microcytosis; target cells frequently are seen on the peripheral smear (Figure 5).7
Sickle cell anemia is an inherited disorder caused by a point mutation leading to a substitution of valine for glutamic acid in the sixth position of the β chain of hemoglobin. Membrane abnormalities from sickling and oxidative damage caused by hemoglobin S, along with impaired deformability of sickle cells, leads to splenic trapping and removal of cells. Some degree of intravascular hemolysis occurs as well. Hemoglobin electrophoresis reveals a predominance of hemoglobin S. Sickle cells are observed on the peripheral smear.
