Glucose-6-phosphate dehydrogenase deficiency, the most common enzyme deficiency worldwide, causes a spectrum of disease including neonatal hyperbilirubinemia, acute hemolysis, and chronic hemolysis. Persons with this condition also may be asymptomatic. This X-linked inherited disorder most commonly affects persons of African, Asian, Mediterranean, or Middle-Eastern descent. Approximately 400 million people are affected worldwide. Homozygotes and heterozygotes can be symptomatic, although the disease typically is more severe in persons who are homozygous for the deficiency. The conversion of nicotinamide adenine dinucleotide phosphate to its reduced form in erythrocytes is the basis of diagnostic testing for the deficiency. This usually is done by fluorescent spot test. Different gene mutations cause different levels of enzyme deficiency, with classes assigned to various degrees of deficiency and disease manifestation. Because acute hemolysis is caused by exposure to an oxidative stressor in the form of an infection, oxidative drug, or fava beans, treatment is geared toward avoidance of these and other stressors. Acute hemolysis is self-limited, but in rare instances it can be severe enough to warrant a blood transfusion. Neonatal hyperbilirubinemia may require treatment with phototherapy or exchange transfusion to prevent kernicterus. The variant that causes chronic hemolysis is uncommon because it is related to sporadic gene mutation rather than the more common inherited gene mutation.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency increases the vulnerability of erythrocytes to oxidative stress. Clinical presentations include acute hemolytic anemia, chronic hemolytic anemia, neonatal hyperbilirubinemia, and an absence of clinical symptoms. The disease is rarely fatal.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Patients with G6PD deficiency should avoid exposure to oxidative drugs (Table 3) and ingestion of fava beans. | C | 3 |
| Neonates should be screened for G6PD deficiency when family history, ethnic or geographic origin, or the timing of the appearance of neonatal jaundice suggests the possibility of G6PD deficiency. | C | 23 |
| G6PD can be diagnosed with a quantitative spectrophotometric analysis or, more commonly, by a rapid fluorescent spot test. | C | 7 |
G6PD = glucose-6-phosphate dehydrogenase.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, see page 1154 orhttps://www.aafp.org/afpsort.xml.
Epidemiology
G6PD deficiency occurs with increased frequency throughout Africa, Asia, the Mediterranean, and the Middle East. In the United States, black males are most commonly affected, with a prevalence of approximately 10 percent. Prevalence of the deficiency is correlated with the geographic distribution of malaria, which has led to the theory that carriers of G6PD deficiency may incur partial protection against malarial infection.1–3 Cases of sporadic gene mutation occur in all populations.
Pathophysiology
G6PD catalyzes nicotinamide adenine dinucleotide phosphate (NADP) to its reduced form, NADPH, in the pentose phosphate pathway (Figure 14). NADPH protects cells from oxidative damage. Because erythrocytes do not generate NADPH in any other way,3 they are more susceptible than other cells to destruction from oxidative stress. The level of G6PD activity in affected erythrocytes generally is lower than in other cells.5 Normal red blood cells that are not under oxidative stress generally exhibit G6PD activity at approximately 2 percent of total capacity.1 Even with enzyme activity that is substantially reduced, there may be few or no clinical symptoms. A total deficiency of G6PD is incompatible with life.6 The G6PD-deficient variants are grouped into different classes corresponding with disease severity(Table 11,7).
Figure 1. Pentose Phosphate Pathway
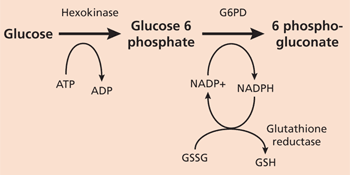
G6PD catalyzes NADP+ to its reduced form, NADPH, in the pentose phosphate pathway. (G6PD = glucose-6-phosphate dehydrogenase; ATP = adenosine triphosphate; ADP = adenosine diphosphate; NADP+ = nicotinamide adenine dinucleotide phosphate [oxidized form]; NADPH = reduced NADP; GSSG = oxidized glutathione; GSH = reduced glutathione.)
Adapted with permission from Glucose 6 phosphate dehydrogenase deficiency. Accessed online July 20, 2005, at:http://www.malariasite.com/malaria/g6pd.htm.
TABLE 1 Classes of G6PD Enzyme Variants
| Class | Level of deficiency | Enzyme activity | Prevalence |
|---|---|---|---|
| I | Severe | Chronic nonspherocytic hemolytic anemia in the presence of normal erythrocyte function | Uncommon; occurs across populations |
| II | Severe | Less than 10 percent of normal | Varies; more common in Asian and Mediterranean populations |
| III | Moderate | 10 to 60 percent of normal | 10 percent of black males in the United States |
| IV | Mild to none | 60 to 150 percent of normal | Rare |
| V | None | Greater than 150 percent of normal | Rare |
G6PD = glucose-6-phosphate dehydrogenase.
Genetics
The gene mutations affecting encoding of G6PD are found on the distal long arm of the X chromosome. More than 400 mutations have been identified, most being missense mutations.6 Most of the variants occur sporadically, although the G6PD Mediterranean and the G6PD A–variants occur with increased frequency in certain populations (Table 23,6,7).
TABLE 2 Comparison of the Two Most Common Variants of G6PD Deficiency
| G6PD Mediterranean | G6PD A– | |
|---|---|---|
| World Health Organization class | Class II | Class III |
| Populations affected | Italian, Grecian, Spanish, Arabic, Jewish (Kurdish) descent | African descent |
| Neonatal hyperbilirubinemia | Yes, may be more severe | Yes |
| Favism | More common | Less common |
| Hemolysis with oxidative drugs | Yes | Yes |
G6PD = glucose-6-phosphate dehydrogenase.
Diagnosis
The diagnosis of G6PD deficiency is made by a quantitative spectrophotometric analysis or, more commonly, by a rapid fluorescent spot test detecting the generation of NADPH from NADP.7 The test is positive if the blood spot fails to fluoresce under ultraviolet light.8 In field research, where quick screening of a large number of patients is needed, other tests have been used; however, they require definitive testing to confirm an abnormal result.9,10 Tests based on polymerase chain reaction detect specific mutations and are used for population screening, family studies, or prenatal diagnosis.6
In patients with acute hemolysis, testing for G6PD deficiency may be falsely negative because older erythrocytes with a higher enzyme deficiency have been hemolyzed. Young erythrocytes and reticulocytes have normal or near-normal enzyme activity. Female heterozygotes may be hard to diagnose because of X-chromosome mosaicism leading to a partial deficiency that will not be detected reliably with screening tests.7,11,12
G6PD deficiency is one of a group of congenital hemolytic anemias, and its diagnosis should be considered in children with a family history of jaundice, anemia, splenomegaly, or cholelithiasis, especially in those of Mediterranean or African ancestry.13
Testing should be considered in children and adults (especially males of African, Mediterranean, or Asian descent) with an acute hemolytic reaction caused by infection, exposure to a known oxidative drug, or ingestion of fava beans.
Although rare, G6PD deficiency should be considered as a cause of any chronic nonspherocytic hemolytic anemia across all population groups.
Newborn screening for G6PD deficiency is not performed routinely in the United States, although it is done in countries with high disease prevalence. The World Health Organization recommends screening all newborns in populations with a prevalence of 3 to 5 percent or more in males.3
Neonatal Hyperbilirubinemia
The prevalence of neonatal hyperbilirubinemia is twice that of the general population14 in males who carry the defective gene and in homozygous females. It rarely occurs in heterozygous females.15,16
The mechanism by which G6PD deficiency causes neonatal hyperbilirubinemia is not completely understood. Although hemolysis may be observed in neonates who have G6PD deficiency and are jaundiced,17 other mechanisms appear to play a more important role in the development of hyperbilirubinemia.6,18,19 Hyperbilirubinemia is likely secondary to impairment of bilirubin conjugation and clearance by the liver leading to indirect hyperbilirubinemia.6,20 Infants with G6PD deficiency and a mutation of uridine diphosphoglucuronate glucuronosyltransferase-1 gene promoter (UDPGT-1) are particularly susceptible to hyperbilirubinemia secondary to decreased liver clearance of bilirubin.21 UDPGT-1 is the enzyme affected in Gilbert disease.
G6PD deficiency should be considered in neonates who develop hyperbilirubinemia within the first 24 hours of life, a history of jaundice in a sibling, bilirubin levels greater than the 95th percentile, and in Asian males.22,23
G6PD deficiency can lead to an increased risk and earlier onset of hyperbilirubinemia,24,25 which may require phototherapy or exchange transfusion.6,25 In certain populations, hyperbilirubinemia secondary to G6PD deficiency results in an increased rate of kernicterus and death,26,27 whereas in other populations this has not been observed.18 This may reflect genetic mutations specific to different ethnic groups.18,19
Acute Hemolysis
Acute hemolysis is caused by infection, ingestion of fava beans, or exposure to an oxidative drug.3 Medications that should be avoided in patients with G6PD deficiency are listed in Table 3,6 and drugs that can be used safely in these patients are listed in Table 4.6 Hemolysis occurs after exposure to the stressor but does not continue despite continued infection or ingestion. This is thought to be a result of older erythrocytes having the greatest enzyme deficiency and undergoing hemolysis first. Once the population of deficient erythrocytes has been hemolyzed, younger erythrocytes and reticulocytes that typically have higher levels of enzyme activity are able to sustain the oxidative damage without hemolysis.7 Clinically, acute hemolysis can cause back or abdominal pain and jaundice secondary to a rise in unconjugated bilirubi n (Table 521). Jaundice, in the setting of normal liver function, typically does not occur until greater than 50 percent of the erythrocytes have been hemolyzed.21
TABLE 3 Medications That Should Be Avoided By Persons with G6PD Deficiency*
| Drug name | Use |
|---|---|
| Dapsone | Antimicrobial for treatment of leprosy |
| Flutamide (Eulexin) | Antiandrogen for treatment of prostate cancer |
| Mafenide cream (Sulfamylon) | Topical antimicrobial |
| Methylene blue (Urolene Blue) | Antidote for druginduced methemoglobinemia |
| Nalidixic acid (NegGram) | Antibiotic used primarily for urinary tract infections |
| Nitrofurantoin (Macrodantin) | Antibiotic used primarily for urinary tract infections |
| Phenazopyridine (Pyridium) | Analgesic for treatment of dysuria |
| Primaquine | Antimalaria agent |
| Rasburicase (Elitek) | Adjunct to antineoplastic agents |
| Sulfacetamide (Klaron) | Antibiotic (ophthalmic and topical preparations) |
| Sulfamethoxazole (Gantanol) | Antibiotic used in combination preparations (i.e., trimethoprim-sulfamethoxazole [TMP-SMX; Bactrim, Septra]) |
| Sulfanilamide (AVC) | Antifungal agent for treatment of vulvovaginalCandida albicans infection |
G6PD = glucose-6-phosphate dehydrogenase.
*—Classes I, II, and III.
Information from reference 6.
TABLE 5 Symptoms and Laboratory Evaluation in Patients with G6PD and Acute Hemolysis
| Symptoms | Laboratory evaluation | Findings in patients with G6PD deficiency and associated acute hemolysis |
|---|---|---|
| Back pain | Complete blood count | Mild to severe anemia |
| Abdominal pain | Reticulocyte count | Increases four to seven days after hemolysis |
| Jaundice | Peripheral blood smear | Heinz bodies |
| Transient splenomegaly | Haptoglobin | Decreased |
| Hemoglobinuria | Liver function tests | Elevated indirect bilirubin |
| Scleral icterus | Coombs’ test | Negative |
G6PD = glucose-6-phosphate dehydrogenase.
Information from references 21.
Drugs that cause hemolysis in G6PD-deficient persons inf lict oxidative damage to erythrocytes leading to erythrocyte destruction. Hemolysis typically occurs 24 to 72 hours after ingestion, with resolution within four to seven days.21 Oxidative drugs ingested by a woman who is breast-feeding may be transmitted in breast milk and can cause acute hemolysis in a G6PD-deficient child.16,28
Although persons who experience hemolysis after the ingestion of fava beans can be presumed to have G6PD deficiency, not all of them will exhibit hemolysis.6,7 Favism is most common in persons with G6PD class II variants, but rarely it can occur in patients with the G6PD A–variant.5 Fava beans (Table 6) are presumed to cause oxidative damage by an unknown component, possibly vicine, convicine, or isouramil.6,7
TABLE 6 Synonyms for Fava Beans
| Bell beans | ||
| Broad beans | ||
| English dwarf beans | ||
| Fever beans | ||
| Haba beans | ||
| Horse beans | ||
| Pigeon beans | ||
| Silkworm beans | ||
| Tick beans |
Infection is the most common cause of acute hemolysis in G6PD-deficient persons,6 although the exact mechanism by which this occurs is unknown. Leukocytes may release oxidants during phagocytosis that cause oxidative stress to the erythrocytes; however, this explanation alone would not account for the variety of infections associated with hemolysis in G6PD-deficient persons. The most common infectious agents causing hemolysis include Salmonella,Escherichia coli, beta-hemolytic streptococci, rickettsial infections, viral hepatitis, and influenza A.
A peripheral smear taken during an acute hemolytic reaction in a G6PD-deficient person may demonstrate Heinz bodies, although this rarely is seen in clinical practice.7
Chronic Hemolysis
In chronic nonspherocytic hemolytic anemia, which usually is caused by a sporadic gene mutation, hemolysis occurs during normal erythrocyte metabolism.5,6 The severity of the hemolysis varies, causing mild hemolysis to transfusion-dependent anemia. Exposure to oxidative stress can cause acute hemolysis in these persons.
Other Clinical Considerations
G6PD-deficient persons are predisposed to the development of sepsis and complications related to sepsis after a severe injury.29 Although research has failed to consistently show a clinically significant risk to patients receiving G6PD-deficient donor blood, blood banks generally do not accept G6PD-deficient blood donors.30
Treatment
The main treatment for G6PD deficiency is avoidance of oxidative stressors. Rarely, anemia may be severe enough to warrant a blood transfusion. Splenectomy generally is not recommended. Folic acid and iron potentially are useful in hemolysis, although G6PD deficiency usually is asymptomatic and the associated hemolysis usually is short-lived. Antioxidants such as vitamin E and selenium have no proven benefit for the treatment of G6PD deficiency.6,31 Research is being done to identify medications that may inhibit oxidative-induced hemolysis of G6PD-deficient red blood cells.32
