Hyperkalemia is a potentially life-threatening metabolic problem caused by inability of the kidneys to excrete potassium, impairment of the mechanisms that move potassium from the circulation into the cells, or a combination of these factors. Acute episodes of hyperkalemia commonly are triggered by the introduction of a medication affecting potassium homeostasis; illness or dehydration also can be triggers. In patients with diabetic nephropathy, hyperkalemia may be caused by the syndrome of hyporeninemic hypoaldosteronism. The presence of typical electrocardiographic changes or a rapid rise in serum potassium indicates that hyperkalemia is potentially life threatening. Urine potassium, creatinine, and osmolarity should be obtained as a first step in determining the cause of hyperkalemia, which directs long-term treatment. Intravenous calcium is effective in reversing electrocardiographic changes and reducing the risk of arrhythmias but does not lower serum potassium. Serum potassium levels can be lowered acutely by using intravenous insulin and glucose, nebulized beta2 agonists, or both. Sodium polystyrene therapy, sometimes with intravenous furosemide and saline, is then initiated to lower total body potassium levels.
The prevalence of hyperkalemia in hospitalized patients is between 1 and 10 percent.1 Although the exact prevalence of hyperkalemia in community-based medical practice is unknown, potassium elevation is a common, potentially life-threatening problem most often occuring in patients with chronic renal failure or other illnesses that reduce renal potassium excretion (Table 12,3). In these patients, acute hyperkalemia often is precipitated by stressors such as illness, dehydration, or initiation of medicines that alter potassium homeostasis (Table 24–10).4–7
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Patients with hyperkalemia who have electrocardiographic (ECG) changes, a rapid rate of rise of serum potassium, decreased renal function, or significant acidosis should be urgently treated. | C | 23 |
| Patients with hyperkalemia and characteristic ECG changes should be given intravenous calcium gluconate. | C | 1–3,27 |
| Acutely lower potassium by giving intravenous insulin with glucose, a beta2 agonist by nebulizer, or both. | C | 2,3,30 |
| Total body potassium should usually be lowered with sodium polystyrene sulfonate (Kayexalate). | C | 2,3,23 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, see page 196 orhttps://www.aafp.org/afpsort.xml.
TABLE 1 Disorders Causing Hyperkalemia
| Disorders leading to hyperkalemia caused by impaired renal excretion of potassium | Disorders leading to hyperkalemia caused by shift of potassium into the extracellular space |
|
|
TABLE 2 Agents That May Cause Hyperkalemia
| Agents | Mechanism and comments |
|---|---|
| Amiloride (Midamor) and triamterene (Dyrenium) | Diminishes potassium secretion by reducing the electrical gradient between the intracellular space and the renal tubule, causing potassium to leave the cells |
| Amino acids* | Lysine, arginine, or epsilon-aminocaproic acid enters cells in exchange for potassium, causing hyperkalemia |
| ARBs and ACE inhibitors | Decreases aldosterone synthesis; hyperkalemia often can be reduced by concomitant diuretic use; ARBs less likely to cause hyperkalemia than ACE inhibitors |
| Azole antifungals | Inhibits adrenal steroid synthesis, which can lead to aldosterone deficiency |
| Beta blockers | Decreases sodium-potassium adenosine triphosphatase (ATPase) activity; beta2 agonists decrease potassium levels |
| Cyclosporine (Sandimmune) | Suppresses renin release, leading to decreased aldosterone synthesis, decreased potassium secretion in collecting duct |
| Digoxin at toxic levels | Decreases sodium-potassium ATPase activity |
| Fluoride toxicity | Decreases aldosterone synthesis; most common in patients on dialysis who drink water with high fluoride levels |
| Glucose infusions or insulin deficiency | Hypertonicity caused by hyperglycemia from glucose infusions can drive potassium out of the intracellular space, leading to hyperkalemia. Hyperkalemia may occur with continuous infusions or with boluses of hypertonic glucose. May be present with hypertonicity caused by other agents such as mannitol (Osmitrol) as well. |
| Heparins | Can cause hyperkalemia in patients with decreased renal function; inhibits adrenal aldosterone synthesis |
| Herbal remedies with digitalis-like effect | Specific agents include milkweed, lily of the valley, Siberian ginseng, Hawthorn berries, or preparations from dried toad skin (Bufo, Chan’su, Senso). All these agents act by decreasing sodium-potassium ATPase activity, leading to elevated extracellular potassium. |
| NSAIDs | Decreased prostaglandin production leads to decreased afferent arteriolar flow, suppressing renin and aldosterone secretion. Typical of NSAIDs as well as cyclooxygenase-2 selective inhibitor drugs. |
| Nutritional and herbal supplements | Herbs containing high potassium levels (e.g., Noni juice, alfalfa, dandelion, horsetail, nettle) |
| Packed red blood cells | Stored cells can partially hemolyze and release potassium when infused. |
| Penicillin G potassium | Can cause hyperkalemia in patients with impaired renal function caused by increased potassium load; can be administered orally or intravenously |
| Potassium supplements or salt substitutes | Ingestion of potassium can lead to hyperkalemia, particularly if renal function is impaired; dietary sources include bananas, melon, and orange juice. |
| Spironolactone (Aldactone) | Inhibits binding of aldosterone to receptors in the renal tubule |
| Succinylcholine (Anectine) | Increases nicotinic acetylcholine receptors in damaged skeletal muscle (e.g., trauma or burn patients) |
| Tacrolimus (Prograf) | Suppresses renin release, leading to decreased aldosterone synthesis and decreased potassium secretion in collecting duct |
| Trimethoprim (Proloprim) and pentamidine (Pentam 300) | Diminishes potassium secretion by reducing the electrical gradient between the intracellular space and the renal tubule, causing potassium to leave the cells. |
ARB = angiotensin receptor blockers; ACE = angiotensin-converting enzymes; NSAID = nonsteroidal anti-inflammatory drug.
*—Hyperkalemia can occur in the setting of amino acids administered intravenously as part of total parenteral nutrition. It is unknown whether oral dietary amino acid supplements cause hyperkalemia.
Normal Potassium Physiology
Two mechanisms normally regulate potassium levels in response to variation of potassium intake. First, ingested potassium rapidly enters the portal circulation, stimulating the pancreas to release insulin. Elevated insulin levels induce rapid transport of potassium from the extracellular space into cells via cellular sodium-potassium adenosine triphosphatase. Second, increased potassium in the circulation causes the renal juxtaglomerular cells to release renin. This stimulates hepatic activation of angiotensin I that is then converted in the lungs to angiotensin II. Angiotensin II stimulates the adrenal zona glomerulosa to secrete aldosterone. Elevated serum aldosterone causes the renal cortical collecting ducts to excrete potassium and retain sodium, further lowering serum potassium.2
Causes of Hyperkalemia
The first step in the evaluation of a patient with elevated serum potassium is to exclude spurious potassium elevation (Table 32,3 ). If the elevation is shown to be real, the next step is to consider: (1) the effects of medications, including increased potassium intake (Table 24–10 ); (2) the impaired distribution of potassium between the intracellular and extracellular space; or (3) the impaired renal excretion of potassium. All three factors often are present (e.g., the stress of illness induces hyperkalemia in a patient rendered susceptible by impaired homeostatic mechanisms and the presence of a medication that impairs normal potassium regulation).
TABLE 3 Causes of Actual Potassium Values That Are Lower Than Laboratory Values
| Drawing blood samples from a vein or line into which potassium is being infused | |
| Laboratory error | |
| Pseudohyperkalemia | |
| Hemolysis | |
| Leukocytosis | |
| Thrombocytosis | |
| Repeated clenching of the fist during phlebotomy | |
| Traumatic venipuncture | |
| Uncommon genetic syndromes | |
| Familial pseudohyperkalemia | |
| Hereditary spherocytosis | |
PSEUDOHYPERKALEMIA
Pseudohyperkalemia occurs when laboratory reports of potassium do not reflect actual values. The most common cause is lysis of red cells in a phlebotomy specimen. Other causes are listed in Table 32,3. Potassium released from platelets can lead to spuriously high levels of potassium in a blood sample allowed to clot to collect serum. Pseudohyperkalemia can be excluded by repeating the sample collection as atraumatically as possible and obtaining serum and plasma potassium levels. In patients with pseudohyperkalemia, the plasma potassium will be normal in the face of an elevated serum potassium.
HYPERKALEMIA CAUSED BY DECREASED EXCRETION OF POTASSIUM
Effective excretion of potassium is dependent on aldosterone and sufficient distal delivery of sodium and water within the nephron. Hyperkalemia may occur when one of these mechanisms is impaired because of renal failure, renal hypoperfusion (e.g., volume depletion, congestive heart failure), or hypoaldosteronism. Hypoaldosteronism may be the cause of hyperkalemia in patients who do not have advanced renal failure or hypoperfusion.11
Hyporeninemic hypoaldosteronism, a syndrome associated with type IV renal tubular acidosis, may be part of the mechanism behind hyperkalemia in patients with mild renal failure, particularly diabetic nephropathy. Hyporeninemic hypoaldosteronism can cause patients who have diabetic nephropathy to develop acute elevations of potassium because of medications or stress (e.g., dehydration, acute illness).12
MEDICATION-INDUCED HYPERKALEMIA
The factors that decrease potassium excretion also increase the risk of medication-induced hyperkalemia. Because of the relative decline in renal function with age, family physicians should use caution when prescribing medications that alter potassium metabolism in older patients. Judicious monitoring of potassium levels is important in at-risk patients receiving these medicines.
Neurohumoral inhibition with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers benefits patients with chronic heart failure; adding spironolactone (Aldactone) reduces morbidity and mortality in patients with severe heart failure.13 ACE inhibition also decreases cardiovascular mortality in high-risk patients, particularly in those with diabetes.14,15 However, patients who take a combination of ACE inhibitors and spironolactone are prone to hyperkalemia8; hyporeninemic hypoaldosteronism may contribute as well. Non-steroidal anti-inflammatory drugs (NSAIDs) decrease renin secretion, leading to decreased potassium secretion. The addition of NSAIDs in these patients can impair renal function to the degree of inducing life-threatening hyperkalemia. Thus, NSAIDs should be taken with caution in patients with diabetes or renal failure.10,16
ADRENAL INSUFFICIENCY
The possibility of adrenal insufficiency should be considered in all patients with hyperkalemia. Clinical suspicion is increased by the presence of hyponatremia and muscular weakness.17 Primary adrenal insufficiency is best screened for with a standard cosyntropin-stimulation test,18 in which 0.25 mg of synthetic cosyntropin is given as an intravenous bolus. Plasma cortisol is measured 45 to 60 minutes later, and values less than 20 mcg per dL (550 nmol per L) suggest adrenal insufficiency.19
CONGENITAL CAUSES OF HYPERKALEMIA
Congenital abnormalities of aldosterone synthesis also can lead to potassium elevation and excessive sodium loss. Severe forms of these disorders lead to electrolyte imbalances in neonates that can be fatal if not corrected promptly. If these patients survive infancy, the disorder tends to be less severe as they get older.20 Pseudohypoaldosteronism refers to congenital resistance to the actions of aldosterone on the kidney; the autosomal recessive form is more severe and can lead to death in the neonate if not treated aggressively. Patients suspected to have one of these unusual genetic abnormalities should be referred to a pediatric endocrinologist to establish appropriate initial treatment; patients may then be managed by their family physician with occasional consultation.
Diagnosis
The initial diagnostic approach begins with the clinical history, review of medications, and physical examination. Symptoms and signs include muscular weakness or flaccid paralysis, ileus, and characteristic electrocardiograph (ECG) changes (Figure 121). Laboratory tests should be directed towards causes suggested by the history and physical examination, with attention to serum electrolytes, creatinine, and blood urea nitrogen. A spot urine test for potassium, creatinine, and osmoles should be obtained to calculate the fractional excretion of potassium and the transtubular potassium gradient (Table 422,23). The transtubular potassium gradient is an assessment of renal potassium handling, with a normal value of eight to nine, rising at times to 11 after an increase in potassium intake. Values lower than five in the face of hyperkalemia suggest an inappropriate renal response to high potassium22; a very low value suggests hypoaldosteronism.
Figure 1.
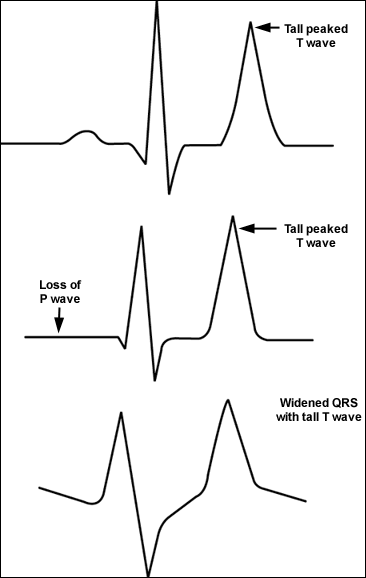
Typical electrocardiograph changes seen in patients with hyperkalemia
Reprinted with permission from Slovis C, Jenkins R. ABC of clinical electrocardiography: conditions not primarily affecting the heart. BMJ 2002;324:1320.
TABLE 4 Diagnostic Equations for Hyperkalemia

| Test* | Formula † | Interpretation in hyperkalemia | Notes | |
|---|---|---|---|---|
| Fractional excretion of potassium (FEK) | FEK less than 10 percent indicates renal etiology FEK greater than 10 percent indicates extrarenal cause | Values can be increased in chronic renal failure. | ||
| or | ||||
| Transtubular potassium gradient | Gradient less than 6 to 8 indicates renal cause Gradient greater than 6 to 8 indicates extrarenal cause. | Values can be increased in chronic renal failure. | ||
| or | ||||
UK = urine potassium; SK = serum potassium; UCr = urine creatinine; SCr = serum creatinine; Uosm = urine osmolality; Sosm = serum osmolality.
*—For the most accurate representation of the kidney’s response to hyperkalemia, these measurements should be drawn before the serum potassium is corrected.
†—Plasma values for potassium and osmolality are recommended for this equation, but serum values are listed because these are more commonly available.
Hyporeninemic hypoaldosteronism should be considered in patients with diabetes and hyperkalemia, who generally have a low serum aldosterone. A trial of oral fludrocortisone (Florinef) is generally the most practical way to empirically establish this diagnosis; if the patient has hyporeninemic hypoaldosteronism, potassium levels will return to normal in a day or two after initiation of fludrocortisone.24
Management
Factors necessitating emergent treatment of hyperkalemia include changes on ECG, a rapid rise of serum potassium, decreased renal function, and the presence of significant acidosis23 (Figure 2). Urgent treatment should not be delayed while a work-up for the etiology is undertaken, although urine potassium, creatinine, and osmolarity studies should be obtained before serum potassium levels are significantly altered. Although controlled trials have not been conducted, it is thought that potassium levels greater than 6.0 mEq per L (6.0 mmol per L) are clinically significant. ECG changes in a patient with hyperkalemia are an ominous portent of potentially fatal arrhythmias. However, hyperkalemia can be life threatening even if the ECG is normal,25,26 and about one half of patients with potassium levels exceeding 6.0 mEq per L have a normal ECG.1
Figure 2. Management of Hyperkalemia
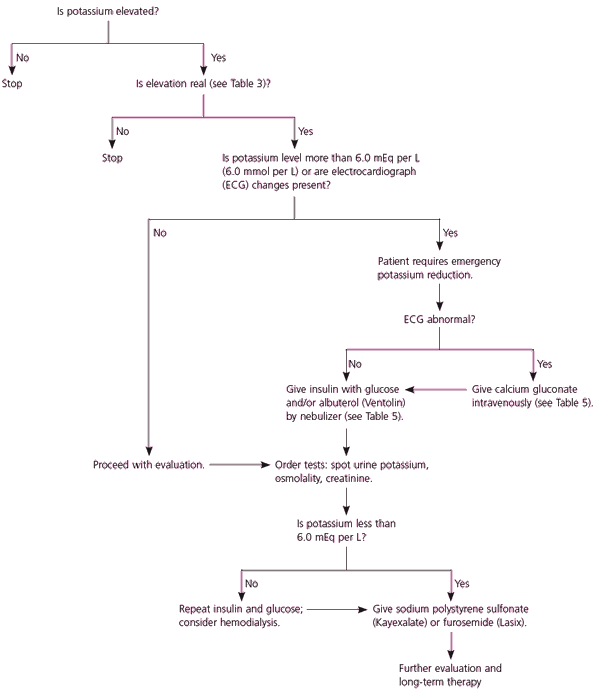
Algorithm for the management of hyperkalemia.
There are no clear guidelines regarding the appropriate setting for the treatment of hyperkalemia. The decision for hospital admission for continuous ECG monitoring is a matter of clinical judgment in each case.2,27 Patients believed to have a rapid rise in potassium commonly need inpatient care, whereas patients whose hyperkalemia has developed over a period of weeks can often be managed in an outpatient setting with close follow-up.
ACUTE TREATMENT
Urgent treatment of hyperkalemia includes stabilizing the myocardium to protect against arrhythmias and shifting potassium from the vascular space into the cells. After the serum potassium level is reduced to safe levels, treatment focuses on lowering total body potassium. In patients who do not require urgent treatment, lowering total body potassium may be the only step necessary.
Intravenous calcium is administered to stabilize the myocardium; it lowers the threshold potential, thus counteracting the toxic effect of high potassium. Calcium does not have any effect on the serum potassium level. Improvement in the ECG changes should be visible within two to three minutes of administration of calcium (Table 52,3). Repeated doses can be given while other measures are initiated.28
TABLE 5 Medications Used in Acute Treatment of Hyperkalemia
| Medication* | Dosage | Onset | Length of effect | Mechanism of action | Cautions |
|---|---|---|---|---|---|
| Calcium gluconate | 10 to 20 mL of 10 percent solution IV over two to three minutes | Immediate | 30 minutes | Protects myocardium from toxic effects of calcium; no effect on serum potassium level | Can worsen digoxin toxicity |
| Insulin | Regular insulin 10 units IV with 50 mL of 50 percent glucose | 15 to 30 minutes | Two to six hours | Shifts potassium out of the vascular space and into the cells; no effect on total body potassium | Consider 5 percent dextrose solution infusion at 100 mL per hour to prevent hypoglycemia with repeated doses. Glucose unnecessary if blood sugar elevated above 250 mg per dL (13.9 mmol per L) |
| Albuterol (Ventolin) | 10 to 20 mg by nebulizer over 10 minutes (use concentrated form, 5 mg per mL) | 15 to 30 minutes | Two to three hours | Shifts potassium into the cells, additive to the effect of insulin; no effect on total body potassium | May cause a brief initial rise in serum potassium |
| Furosemide (Lasix) | 20 to 40 mg IV, give with saline if volume depletion is a concern | 15 minutes to one hour | Four hours | Increases renal excretion of potassium | Only effective if adequate renal response to loop diuretic |
| Sodium polystyrene sulfonate (Kayexalate) | Oral: 50 g in 30 mL of sorbitol solution Rectal: 50 g in a retention enema | One to two hours (rectal route is faster) | Four to six hours | Removes potassium from the gut in exchange for sodium | Sorbitol may be associated with bowel necrosis. May lead to sodium retention |
IV = intravenously.
*—Medications listed in order of use from most to least urgent.
Caution should be used in patients who take digoxin because calcium has been reported to worsen the myocardial effects of digoxin toxicity.2,3 Some experts suggest using a slower calcium infusion for 20 to 30 minutes in patients with hyperkalemia who are on digitalis therapy.28–30 An alternative is to consider using magnesium instead of calcium to stabilize the myocardium.29
Shifting potassium intracellularly is done using insulin or a beta2 agonist (Table 52,3). Insulin typically is given as 10 units intravenously with 50 mL of 50 percent glucose to counteract hypoglycemia. Repeated doses can be given if the potassium level remains elevated.
Inhaled beta2 agonists have a rapid onset of action. The effect of beta2 agonists is additive to that of insulin administration, and they can be taken together.31 Nebulized albuterol (Ventolin) is taken in a dose of 10 to 20 mg. Intravenous beta2 agonists have been used in Europe, but they are not approved by the U.S. Food and Drug Administration.3
Sodium bicarbonate is no longer recommended to lower potassium, although it may be appropriate in patients with severe metabolic acidosis.32
LOWERING TOTAL BODY POTASSIUM
Treatments that shift potassium into the cells have no effect on total body potassium. Potassium can be eliminated by renal excretion, gastrointestinal elimination, or dialysis. The agents taken to lower total body potassium can interfere with tests to determine the cause of hyperkalemia. Thus, spot urine potassium, creatinine, and osmolality levels should be obtained before the agents are initiated; however, treatment should not be delayed while awaiting results.
Gastrointestinal excretion is accomplished using sodium polystyrene sulfonate (Kayexalate), which binds potassium in the colon in exchange for sodium; it can be given orally or as a retention enema. The enema form is faster; the oral route can take four to six hours because it requires the resin to get to the colon before it takes effect. Sodium polystyrene sulfonate often is given with sorbitol to decrease constipation. However, sorbitol can have intestinal complications, with reports of bowel necrosis and perforation in immunocompromised patients.33 Using furosemide (Lasix) with polystyrene reduces the risk of volume overload because of the sodium that is exchanged for potassium by the resin (Table 52,3).33
Excretion of renal potassium can be increased with the use of diuretics, particularly loop diuretics (e.g., furosemide). Patients with decreased kidney function may be relatively resistant to the effects of diuretics. If the patient is volume depleted, saline can be given with the diuretic. Hemodialysis or continuous renal replacement therapy are the treatments of last resort, with the exception of patients already receiving these therapies.
LONG-TERM TREATMENT
Long-term treatment should be tailored to correcting the underlying cause of hyperkalemia. Low-potassium diets should be discussed with patients, and medications that precipitated hyperkalemia should be discontinued if possible. The use of loop diuretics or fludrocortisone will be needed for patients with hyporeninemic hypoaldosteronism whose hyperkalemia recurs or is chronic. The usual dosage of f ludrocortisone is 0.1 mg daily, although more will be needed in some patients. In some patients, hyporeninemic hypoaldosteronism is transitory and resolves after acute management; in others, long-term management with f ludrocortisone is required. Many patients tolerate long-term use of f ludrocortisone with no problems. The principal side effects are hypertension and f luid retention, which may respond to an added diuretic. Although the question of appropriate treatment duration with f ludrocortisone has never been studied, one approach to management would be to slowly taper f ludrocortisone on an outpatient basis, and reinstate f ludrocortisone if potassium rises.
Hyperkalemia caused by the use of ACE inhibitors or angiotensin receptor blockers in patients with chronic renal failure and metabolic acidosis may respond to sodium bicarbonate supplementation. The dosage is 25 to 50 mEq daily (two tablets twice a day at 8 mEq each,) or baking soda (1/2 to 1 tsp daily). Concomitant diuretic use limits the risk of volume overload.12
