Retinoblastoma, a neuroblastic tumor, is the most common primary intraocular malignancy of childhood. Patients usually present with leukokoria (white reflex or white pupil), detected in primary care. The mean age at diagnosis is 12 months for bilateral tumors and 24 months for unilateral tumors. If untreated, almost all patients die of intracranial extension and disseminated disease within two years. However, new diagnostic and treatment methods allow for a high cure rate (93 percent five-year survival in the United States), therefore it is important that primary care physicians recognize the manifestations of this malignancy. Diagnosis is primarily by history and complete ophthalmic examination, with studies including ultrasonography of the eye and imaging of the orbits and brain. Treatment modalities include laser thermotherapy, cryotherapy, radioactive plaques, external beam radiotherapy, chemotherapy, and enucleation. Prospective parents with a family history of retinoblastoma should be referred for genetic counseling. Office evaluation for a red reflex in children should be performed until three years of age. If leukokoria is observed, the patient should be examined by an ophthalmologist within one week.
Retinoblastoma is a neuroblastic tumor that usually is detected in primary care. It is the most common primary intraocular malignancy of childhood, with an incidence of approximately one in 20,000, and affects males and females equally.1 The median age at diagnosis is 12 months in those with bilateral tumor and 24 months in those with unilateral disease.1 Onset after five years of age is rare, but retinoblastoma has been reported in adults.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Prospective parents with a family history of retinoblastoma should be referred for genetic counseling. | C | 3 |
| All children with a family history of retinoblastoma should be screened soon after birth and then at scheduled intervals. | C | 4 |
| Children with leukokoria (white reflex) should be examined by an ophthalmologist within one week. | C | 4 |
| Patients with a history of retinoblastoma should be examined annually for secondary malignancies. | C | 17,19,21 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, see page 956 orhttps://www.aafp.org/afpsort.xml.
Natural History
Untreated, retinoblastoma will grow and produce seeding in the eye, leading to retinal detachment, necrosis and invasion of the orbit, optic nerve invasion, and central nervous system invasion. Metastases generally occur within 12 months. Most commonly, metastases occur through direct invasion of the central nervous system via the optic nerve. The tumor also may spread through the subarachnoid space to the contralateral optic nerve or through the cerebrospinal fluid to the central nervous system, as well as hematogenously to the lungs, bone, and brain. Almost all untreated patients die of intracranial extension and disseminated disease within two years. Poor prognostic factors include a delay in tumor diagnosis, larger tumor, greater age, evidence of optic nerve involvement, and extraocular extension.2
Genetics
The retinoblastoma gene is a tumor suppressor gene, located on the long arm of chromosome 13 at region 14, that codes for the RB protein.3 Disease occurs from any mutation that inactivates both normal alleles. Approximately 60 percent of retinoblastoma occurrences are secondary to somatic, non-hereditary mutations. Such mutations result in predominantly unilateral, unifocal tumors. Approximately 40 percent of tumors result from germline mutations, either inheritance of a preexisting familial germline mutation (positive family history, 10 percent) or a new-onset germline mutation (negative family history, 30 percent). The typical inheritance pattern is of autosomal dominant type.
Diagnosis
Leukokoria (white reflex, or white pupil, instead of the normal red reflex) is the most common presenting sign and often is noticed by the family (Figure 1).4 On physical examination, normal red reflex is more orange than red (the term is misleading), and can vary by iris pigmentation. Depending on the angulation, even the normal optic disc can cast a yellowish hue, and this should not be cause for alarm. An abnormal red reflex is marked by a lack of red-orange reflex, which may or may not be replaced by a white reflex (from the white-colored tumor), as in leukokoria, and is consistently absent in all positions or in a certain position of gaze.
Figure 1.
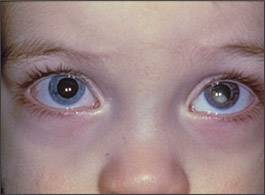
A two-year-old with leukokoria in the left eye.
At well-child visits from birth to three years of age, parents should be asked whether they have any concerns about their child’s eyes. Physical examination in the nursery and the office also should include evaluation for a red reflex and for any ocular abnormalities until a child reaches three years of age, after which visual acuity can be tested. If leukokoria is observed, or if there is any doubt about the red reflex, the child should see an ophthalmologist within one week.
The second most common sign of retinoblastoma is strabismus; other possible manifestations include red eye, tearing, corneal clouding, discoloration of the iris (caused by neovascularization), inflammation, hyphema (blood in the anterior chamber), and glaucoma.4
Rarely, a tumor with features similar to retinoblastoma develops in the parasellar region of the brain or in the pineal gland (pinealoblastoma).5 Occurrence of pinealoblastoma in combination with bilateral retinoblastoma, also known as trilateral retinoblastoma, is observed in 8 percent of patients with bilateral or familial retinoblastoma. The diagnosis of trilateral retinoblastoma usually is made within one year of diagnosis of retinoblastoma.6 Patients with trilateral retinoblastoma may present with headache, vomiting, hydrocephalus, and meningismus.
Diagnosis of retinoblastoma usually is based on the ophthalmoscopic appearance of the tumor, but a complete history and review of systems is always indicated. Particular attention should be paid to risk factors for other diseases that present with leukokoria. A differential diagnosis of leukokoria is given in Table 1.7
TABLE 1 Differential Diagnosis of Leukokoria
| Diagnosis | Pathophysiology |
|---|---|
| Cataract | Opacification of the lens |
| Coats’ disease | Accumulation of subretinal fluid and lipid |
| Primary persistent hyperplastic vitreous | Remnants of embryologic mesenchymal tissue in vitreous cavity |
| Retinal detachment | Fluid under the retina |
| Retinoblastoma | Ocular tumor |
| Retinopathy of prematurity | Abnormal retinal development |
| Toxocariasis | Retinal granulomas |
| Other congenital, degenerative, and inflammatory diseases of the retina | Varies |
Information from reference 7.
DIAGNOSTIC TESTING
Ultrasonography is helpful in detecting intralesional calcification, which is typical of retinoblastoma (Figure 2). Computed tomography also can detect calcification, but it is used less often because of concerns about radiation exposure. Pretreatment investigative studies also should include magnetic resonance imaging (with contrast) of the orbits and brain to look for extraocular extension and to exclude trilateral retinoblastoma in patients with germline mutations.
Figure 2.
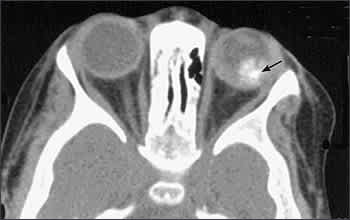
Metastatic disease with no evidence of optic nerve involvement is rare. Lumbar puncture with cerebrospinal fluid analysis, bone marrow aspiration, and bone scan are not performed routinely. However, these tests are indicated in children with advanced intraocular disease or with evidence of extra-ocular disease at presentation.8
Family History
Prospective parents with a family history of retinoblastoma should be referred for genetic counseling. All children with a family history of retinoblastoma should be screened soon after birth; screening should be repeated every four to six weeks until one year of age, and then every two to three months until three years of age. The risk of retinoblastoma is variable depending on the relationship to the affected family member and the type of mutation. Consideration should be given to RB gene mutation testing in relatives who are at risk.3
In a retrospective study9 of 1,831 patients with retinoblastoma, those with a family history of retinoblastoma who were monitored from birth were diagnosed at a younger age (average eight months) and had better five-year ocular survival than those who had a positive family history and did not undergo surveillance (68 versus 38 percent, respectively, with 71 versus 15 percent ocular survival for unilateral tumors and 67 versus 43 percent for bilateral tumors).
Differential Diagnosis
Most diseases can be differentiated readily from retinoblastoma by a careful clinical examination (Table 1).7 Coats’ disease is an idiopathic eye disease that occurs predominantly in boys. It is characterized by pathologic telangiectatic retinal vessels that leak and lead to accumulation of subretinal fluid and lipid, which appears as leukokoria. Coats’ disease is the most common condition mistaken for retinoblastoma, but it is differentiated by a lack of calcification of the retinal mass. Primary persistent hyperplastic vitreous, also known as persistent fetal vasculature syndrome, is a congenital unilateral developmental anomaly characterized by persistence of embryologic mesenchymal tissue remnants in the vitreous cavity. Patients often present with leukokoria; however, there is no retinal mass present in primary persistent hyperplastic vitreous.
Congenital cataracts are an important cause of childhood leukokoria. They often are present at birth and can be idiopathic, familial, or associated with a number of childhood or maternal diseases such as rubella, syphilis, and galactosemia. Careful examination with the slit lamp will identify cataracts. Toxocara infection causes retinochoroidal scarring and vitreous inflammation; this distorts normal retinal architecture and can manifest as leukokoria on ophthalmoscopy. Serum enzyme-linked immunosorbent assay for Toxocara canis can be used to confirm the diagnosis. Retinopathy of prematurity results from the failure of development of the normal retina in premature neonates exposed to high levels of oxygen during the postnatal period. It is associated with abnormal vascularization, fibrosis, and retinal detachment, which can produce a white reflex, and should be suspected in children born prematurely.
Treatment
The treatment of retinoblastoma has changed dramatically in recent years owing to the evolution of treatment options (Table 2).10–16 The goals of treatment are primarily to save the patient’s life and secondarily to save the eye. Treatment options include laser thermotherapy, cryotherapy, radioactive plaques, external beam radiotherapy, chemotherapy, and enucleation. Choice of treatment is determined largely by the size, location, and laterality of the tumor and the visual potential and age of the child (Figure 3).
TABLE 2 Treatment Options for Retinoblastoma
| Treatment | Indications | Complications | Cure rate (%) |
|---|---|---|---|
| Laser thermotherapy11 | One or a few small tumors with no vitreous seeding | Tumor recurrence, retinal detachment, retinal vascular occlusion, preretinal fibrosis | Up to 92 |
| Cryotherapy12 | One or a few small tumors with no vitreous seeding; more suitable for anterior tumors | Retinal breaks, retinal detachment, choroidal detachment, uveitis | 70 or more |
| Radioactive plaques13 | Tumors too large to treat with thermotherapy or cryotherapy | Optic neuropathy, radiation retinopathy | Up to 90 |
| External beam radiotherapy14 | Large bilateral tumors that have failed chemotherapy; tumors near the fovea or optic nerve; tumors too large for thermotherapy or cryotherapy; should be avoided in patients younger than 12 months | Optic neuropathy, radiation retinopathy, cataracts, dry eye, muscle atrophy; high risk of secondary malignant tumors | Up to 90 |
| Chemotherapy15 | Suspected or documented metastatic disease; to reduce the size of a tumor in preparation for local therapy | Bone marrow suppression, nephropathy, deafness, possible leukemia | 50 to 90 |
| Enucleation16 | Unilateral, large tumors that occupy 50 percent or more of the intraocular volume, that demonstrate anterior segment extension or neovascular glaucoma, or that fail to respond to more conservative therapy | Generally safe; rare complications include postoperative hemorrhage and infection | 95 |
Figure 3.
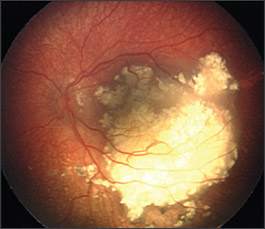
Fundus photograph showing a large retinoblastoma that has regressed into a calcific mass following chemotherapy.
Ophthalmic Follow-up
Up to 45 percent of patients treated with eye-preserving therapy may need subsequent therapy for recurrence or for a new ocular tumor.17 Up to 10 percent of patients with unilateral tumors will develop tumor in the contralateral eye.18
Second Malignant Neoplasm
Fifty percent of patients with germline mutations develop a second, nonocular malignant neoplasm by 50 years of age (Figure 4).19 The most common types are osteosarcoma, soft tissue sarcoma, and cutaneous melanoma,20 although in the first few years following initial diagnosis the most common malignancy is trilateral retinoblastoma. External beam radiotherapy is associated with an increased risk of malignancy in the irradiated area relative to the dose of irradiation.19 The incidence of malignancy is greatest when radiotherapy is used in patients younger than 12 months.21 Patients should be counseled on the signs and symptoms of second tumors and should have a complete examination yearly.
Figure 4.
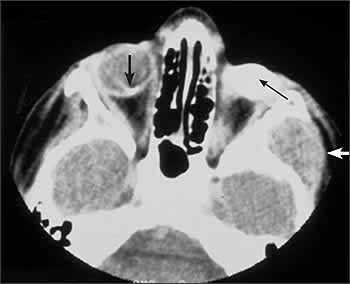
Computed tomography (CT) scan of a 15-year-old girl who presented with a mass of the left temple region. She had undergone enucleation of the left eye and external beam radiotherapy of both eyes for bilateral retinoblastoma at 18 months of age. The CT scan shows a prosthesis in the left socket (thin black arrow), calcified regressed retinoblastoma in the right eye (thick black arrow), and the temporal fossa mass (white arrow). Subsequent biopsy of the temporal fossa mass revealed osteogenic sarcoma.
