Sickle cell disease represents a spectrum of inherited hemoglobin disorders. The pathophysiology involves abnormalities not just in red blood cells but also vascular endothelium, white blood cell function, coagulation, and inflammatory response. Known sequelae of sickle cell disease include invasive infections, painful episodes, acute chest syndrome, strokes, and chronic pulmonary hypertension. Preventive strategies that decrease the risk of infection are the routine use of daily antibiotics until five years of age, immunization of children with the 7-valent pneumococcal conjugate vaccine in addition to the 23-valent polysaccharide pneumococcal vaccine, annual influenza vaccination after six months of age, and meningococcal vaccination after two years of age. A significant advance in stroke prevention is the use of transcranial Doppler ultrasonography to identify asymptomatic, at-risk children who should be considered for chronic blood transfusions. Chronic transfusion therapy for primary or secondary stroke prevention requires careful surveillance for iron overload and chelation therapy. Patients with chest pain, fever, or respiratory symptoms and new pulmonary infiltrates require aggressive medical management for acute chest syndrome. Pain management still represents an important area for aggressive treatment using sickle cell disease–specific guidelines. Newer treatments include hydroxyurea therapy to decrease the frequency of painful episodes and associated comorbidities, and hematopoietic cell transplantation for a limited subset of patients. Family physicians play a crucial role in instituting evidence-based preventive care strategies, initiating timely treatment of acute illness, recognizing life-threatening episodes, and providing a medical home for multidisciplinary management.
In just one generation, the average survival of patients with sickle cell anemia (hemoglobin SS) has increased from 14 years to nearly 50 years.1,2 Sickle cell disease includes a number of genotypes (Figures 1 and 2) and a wide range of associated phenotypes characterized by chronic hemolysis, tissue infarction, and painful episodes (Table 1). Sickle cell disease was once thought to involve solely the polymerization of hemoglobin S, but it is now known that vaso-occlusion also involves leukocytes, activated vascular endothelium, altered nitric oxide metabolism, hypercoagulability, and ischemia-reperfusion injury.3,4 Advances in the understanding of sickle cell disease patho-physiology have led to new opportunities to improve patient care. This article provides an update of prevention and management strategies to modify the progression of disease and enable primary care physicians to better recognize complications that require the input of subspecialists.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Screening | ||
| Primary care physicians should screen infants and children for hemoglobinopathies if no records are available. | B | 5, 6 |
| Children with sickle cell disease should be screened with transcranial Doppler ultrasonography starting at two years of age (frequency of rescreening has not been established). | C | 18, 19 |
| Recommended frequency of comprehensive medical examination should be guided by ongoing consultation with hematologists specializing in sickle cell disease. Physicians should screen for asymptomatic renal, pulmonary, and liver disease, and for drug-related complications for specific therapies. | C | 10, 28 |
| Patients with sickle cell disease should receive periodic retinal screening beginning at 10 years of age. | C | 10 |
| Vaccines for patients with sickle cell disease* | ||
| In patients six months and older, influenza vaccine should be given annually. | C | 14 |
| The 7-valent pneumococcal conjugate vaccine (Prevnar) should be administered as for children without sickle cell disease. | B | 12 |
| The 23-valent polysaccharide pneumococcal vaccine should be administered if two years or older, with another dose at three to five years (if patient is 10 years or younger) or five years (if patient is older than 10 years) after the first dose. | B | 12 |
| Meningococcal vaccine is recommended for patients two years and older with sickle cell disease. | C | 15 |
| Interventions | ||
| Infants and children with sickle cell disease should receive prophylactic daily penicillin V potassium (Veetids) starting by two months of age. Dosages change at three years of age. | A | 7, 8 |
| Antibiotic prophylaxis may be stopped after five years of age for patients without a history of invasive pneumococcal infection or splenectomy. | B | 9 |
| Hydroxyurea should be considered for adults and older adolescents with sickle cell anemia who experience three or more moderate to severe vasoocclusive painful episodes per year. | A | 34 |
| Hydroxyurea should be considered for adults with a history of severe or recurrent acute chest syndrome or symptomatic anemia. | C | 28 |
| Hospital-based treatment of acute painful episodes | ||
| Optimize pain management using opioids based on American Pain Society algorithms. | C | 27 |
| Intravenous fluids should be hypotonic and, after correction for volume depletion, limited to maintenance requirements. | C | 29 |
| Incentive spirometry should be used during waking hours. | A | 30 |
| Monitor oxygen saturation and avoid oversedation. | C | 29 |
*—Updated immunization schedules for children and adults, including when vaccine status is unknown, for catch-up immunizations, and for special situations, are available athttp://www.cdc.gov/nip/recs/child-schedule.htm#mmwr andhttp://www.cdc.gov/nip/recs/adult-schedule.pdf.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, see page 215 orhttps://www.aafp.org/afpsort.xml.
Figure 1.
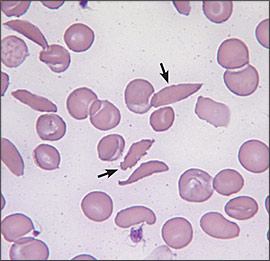
Peripheral blood smear of a patient with hemoglobin SS disease. Arrows indicate classic sickle cells.
Figure 2.
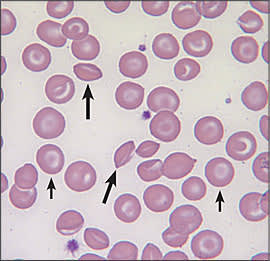
Peripheral blood smear of a patient with hemoglobin SC disease. Short arrows indicate target cells; long arrows indicate atypical sickle cells.
TABLE 1 Findings in Patients with Sickle Cell Syndromes After Five Years of Age
| Hemoglobinopathy | Clinical manifestations | Hb (g per dL) | Hb S (%) | Hb A (%) | Hb A2(%) | Hb F (%) |
|---|---|---|---|---|---|---|
| Sickle cell trait (carrier) | Asymptomatic* | Normal | 40 | 60 | < 3.5 | 0 |
| Sickle cell disease (common variants) | ||||||
| Hb SS (sickle cell anemia) | Usually marked † | 6 to 8 | > 90 | 0 | < 3.5 | < 10 |
| Hb S beta°-thalassemia | Marked to moderate † | 7 to 9 | > 80 | 0 | > 3.5 | < 20 |
| Hb S beta+-thalassemia | Mild to moderate † | 9 to 12 | > 60 | 10 to 30 | > 3.5 | < 20 |
| Hb SC | Mild to moderate † | 10 to 15 | 50 | 0 | ‡ | 0 |
Hb = hemoglobin.
*—Considered a benign condition. Reported complications include isosthenuria, hematuria, splenic infarction at high altitude, and, rarely, sudden death associated with extreme dehydration and hyperthermia.
†—Individual variability.
‡—Hb C 50 percent.
Integrating New Preventive Care Strategies
Neonatal diagnostic screening for hemoglobinopathies, timely institution of family education, and antibiotic prophylaxis remain among the most successful preventive care strategies. Not all states have universal screening, and screening may not have been available for children born in another country; therefore, if no record is available, physicians should consider screening new patients.5,6 Regarding antibiotic prophylaxis, infants and children with sickle cell disease should receive prophylactic daily penicillin V potassium (Veetids) starting by two months of age (intramuscular penicillin G benzathine [Bicillin L-A] given every three weeks can be substituted).7,8 Antibiotic prophylaxis can be stopped safely at five years of age for patients without a history of invasive pneumococcal infection or splenectomy.9 Primary care physicians can improve outcomes by augmenting vaccinations based on risk, screening children to identify those who will benefit from stroke prophylaxis, and screening for pulmonary hypertension. There is a need for periodic comprehensive medical examinations that include assessment of liver, pulmonary, and renal functions. The frequency of examinations should be guided by consultation with hematologists specializing in sickle cell disease. Ophthalmologic examination should be performed periodically after the age of 10.10
SPECIAL RECOMMENDATIONS FOR IMMUNIZATION
Children with sickle cell disease should receive all routine childhood immunizations. The new 7-valent pneumococcal conjugate vaccine (PCV; Prevnar) decreases the incidence of invasive pneumococcal infection in children younger than two years and should be given to infants with sickle cell disease on the same schedule as other children.11,12 However, because children with sickle cell disease remain susceptible to life-threatening pneumococcal infections, they should receive the 23-valent polysaccharide pneumococcal vaccine (PPV) if two years or older; and, if the patient is 10 years or younger, revaccination should be considered three to five years later.12,13 These immunizations do not obviate the need for penicillin prophylaxis, which should continue until five years of age.8 Other recommendations include annual influenza immunization for patients six months and older and meningococcal vaccination for patients two years and older.14,15
ADDRESSING THE HIGH INCIDENCE OF STROKE
Neurologic complications are relatively common in children and adults with sickle cell disease.16 In fact, 17 percent of asymptomatic children with hemoglobin SS disease have structural changes on magnetic resonance imaging consistent with “silent” infarctions.17 Occlusive strokes are attributed to persistent sickling and probable endothelial proliferation, leading to narrowing of cerebral blood vessels. Fortunately, primary prevention of childhood stroke is now possible because effective screening and treatment exist.
Screening with specialized transcranial Doppler ultrasonography is recommended for asymptomatic children with sickle cell disease beginning at two years of age to identify those with an abnormal transcranial blood-flow velocity (TBV).18,19 Children with TBV of 200 cm per second or more are at a particularly high risk of stroke.20 In a multicenter clinical trial18 using this TBV criterion for enrollment, 16.4 percent of children receiving standard care had a stroke during a mean follow-up of 18 months; chronic transfusions reduced the incidence of stroke to 1.6 percent. Furthermore, the most recent data indicate that patients revert back to high-risk status if the transfusions are discontinued.21 Transfusion therapy should be considered for children at increased risk of stroke.19
Recognition of incipient stroke is critical for all children and adults. Symptoms may be as subtle as behavioral changes or a prolonged headache, or even may be part of an uncomplicated painful episode. Prompt brain imaging studies (computed tomography and magnetic resonance angiography) should be performed, and consultation with neurology and hematology subspecialists should be obtained if there is any suspicion for stroke.
EARLY RECOGNITION OF PULMONARY HYPERTENSION
The prevalence of pulmonary hypertension in adults with sickle cell disease may be as high as 30 percent.22,23 Children may be affected as well. Transthoracic Doppler echocardiography with attention to the tricuspid regurgitant jet can identify a subset of patients at increased risk of death (odds ratio, 10.1).24 Thus, a high index of suspicion is warranted when evaluating patients with sickle cell disease who have unexplained dyspnea, hypoxemia, physical examination findings such as increased intensity of the second heart sound, or right ventricular enlargement on chest radiography. Many experts now advocate periodic screening of all adults with sickle cell disease. Echocardiography should be performed when the patient is not acutely ill; those who have abnormal study results should then be referred to a hematologist. Treatment of pulmonary hypertension associated with hemolytic anemias is an area of active investigation at many referral centers.
Management of Acute Complications
PAINFUL EPISODES
Painful episodes occur when sickled erythrocytes cause microvascular occlusion that results in tissue inflammation and ischemia. Comprehensive approaches to ambulatory and inpatient pain management have been addressed in a number of publications, including two with useful algorithms.25,26 In addition, monographs published by the American Pain Society27 and the National Institutes of Health28 are of particular benefit in establishing clinical pathways for hospital-based pain management in patients with sickle cell disease.
Recommendations include the timely use of opioid medications for moderate to severe pain and avoidance of meperidine (Demerol). Oxygen supplementation is not needed unless hypoxemia is present. Close monitoring of oxygen saturation and respiratory status, with particular attention to avoid excessive sedation, is necessary.29 Ancillary measures to reduce the incidence of complications include the use of frequent incentive spirometry while the patient is awake and avoidance of fluid overload by limiting overall intake to 1.0 to 1.5 times maintenance needs.29,30
ACUTE CHEST SYNDROME
Acute chest syndrome, characterized by respiratory or chest symptoms, fever, and a new pulmonary infiltrate on chest radiography, has many etiologies (Table 2).29 Acute chest syndrome may complicate any painful episode or other acute medical condition in a patient with sickle cell disease. For example, 30 percent of all patients with hemoglobin SS disease will have one episode of acute chest syndrome; one half of these patients will have recurrent episodes.31 Because survival is improved with aggressive treatment, urgent consultation with a hematologist or pulmonologist is advised. For persistent hypoxemia or respiratory deterioration in the face of conservative measures, transfusion of red blood cells is indicated.29
TABLE 2 Acute Chest Syndrome: Presentation and Management
| Symptoms | |
| New onset of chest pain | |
| Fever | |
| Wheezing | |
| Dyspnea or tachypnea | |
| Cough | |
| Chest radiography findings | |
| New pulmonary infiltrate | |
| Clinical setting | |
| As the presenting condition without apparent precipitating factors | |
| During painful episodes | |
| During neurologic events | |
| Active respiratory infection (e.g., bronchitis, pneumonia) | |
| Postoperative | |
| Etiology | |
| Fat embolism | |
| Infection | |
| Bacteria (including mycoplasma, legionella, chlamydia) | |
| Mixed infection | |
| Viruses | |
| Pulmonary infarction | |
| Laboratory testing | |
| Blood and sputum cultures | |
| Continuous or interval pulse oximetry | |
| Daily complete blood count and serum chemistry panel | |
| Arterial blood gas measurement for change in oxygen saturation | |
| Pre- and post-transfusion hemoglobin S concentration (hemoglobin electrophoresis) | |
| Treatment | |
| Supplemental oxygen for Po2 below 80 mm Hg or oxygen saturation below 95 percent | |
| Incentive spirometry while awake | |
| Broad-spectrum antibiotic therapy to include coverage for atypical organisms | |
| Fluid management with attention to cardiopulmonary status, daily intake/output analysis, and daily weight | |
| Aggressive pain management with respiratory monitoring and avoidance of oversedation | |
| Bronchodilator therapy if reactive airway disease | |
| Transfusion therapy for hypoxemia or clinical deterioration occurring despite the above interventions | |
Po2 = oxygen partial pressure.
Information from reference 29.
MULTIPLE ORGAN FAILURE SYNDROME
This life-threatening complication involves the acute development of severe dysfunction in at least two of three major organs (kidney, liver, or lung) during a painful episode. Fever, a drop in hemoglobin level, decreasing platelet count, rhabdomyolysis, and mental status changes are all associated with the onset of multi-organ failure. This rare complication is one of the most challenging for even the most experienced sickle cell disease expert. Prompt consultation with a hematologist is advised because emergent exchange transfusion can be lifesaving.32
Strategies to Modify Disease Outcomes
HYDROXYUREA THERAPY
Hydroxyurea increases hemoglobin F levels, improves red cell deformability and hydration, and reduces red cell adhesion to vascular endothelium.33 In a randomized placebo-controlled trial, adults with hemoglobin SS receiving hydroxyurea had significant reductions in painful crises (from 4.5 to 2.5 per year), the incidence of acute chest syndrome, and the frequency of transfusions.34 Follow-up data at nine years from an observational study35 of patients on hydroxyurea showed not only continuing clinical response but also a 40 percent reduction in mortality. Thus, adults and older adolescents with sickle cell disease who experience frequent painful episodes or who have a history of severe or recurrent acute chest syndrome, other vaso-occlusive episodes, or symptomatic anemia should be considered candidates for hydroxyurea therapy.28 Table 3 shows the types of assessment, baseline work-up, and laboratory studies required by the consulting hematologist for treatment initiation.
TABLE 3 Hydroxyurea Therapy in Sickle Cell Disease: An Overview
| Baseline laboratory evaluation | |
|---|---|
| CBC with WBC differential and reticulocyte count | |
| Hemoglobin F measurement | |
| Renal and liver function tests | |
| Pregnancy test | |
| Counseling | |
| Women and men taking hydroxyurea should use contraception. | |
| Dosage modification is required to optimize opportunity for clinical response and avoid complications from myelotoxicity. | |
| Monitoring | |
| Blood counts at frequent intervals early in therapy and when dosage changes | |
| Periodic renal and liver function tests | |
| Hemoglobin F levels and red blood cell MCV values to detect a biologic response (i.e., an increase in these parameters) | |
| Patient compliance | |
| Dosage escalation end points | |
| Reduction in sickle cell pain frequency and/or severity | |
| A treatment period of six months may be needed to assess response in patients who continue to have symptoms. | |
| If lack of clinical response: (1) myelotoxicity that defines the maximum tolerated dosage for the patientor (2) maximum dosage of 35 mg per kg per day | |
CBC = complete blood count; WBC = white blood cell; MCV = mean corpuscular volume.
Comanagement of chronic hydroxyurea therapy requires the family physician and office personnel to be familiar with the side effects of the medication and to be able to provide close follow-up during therapy. Clinical studies of hydroxyurea treatment in infants and children show promising results as well, but this therapy is not approved by the U.S. Food and Drug Administration for this population, and longer-term follow-up is necessary to ascertain the risk of adverse effects.36
ACUTE AND CHRONIC TRANSFUSION THERAPY
Family physicians should request consultation with a hematologist and a blood bank physician when considering transfusions for patients with sickle cell disease. In general, transfusions should not be used for uncomplicated, acute, painful episodes; minor surgical procedures; chronic anemia without symptoms; or uncomplicated pregnancies. As described earlier, transfusion therapy often is of great benefit. The risks of transfusion therapy, whether acute or chronic, need to be managed carefully to avoid adverse events, because the end points for transfusion in patients with sickle cell disease differ from the end points in other patients.
Exchange transfusion may be necessary as an alternative to simple transfusion to avoid hyperviscosity and volume overload. The blood bank should be notified immediately of the possible need for transfusion because patients with sickle cell disease have special needs for matching red blood cell antigens, and they are at increased risk for transfusion reactions because of sensitization by previous transfusions. Delayed hemolytic transfusion reactions may be difficult to recognize because the symptoms may overlap those of a severe painful episode. Finally, patients who are receiving chronic transfusions must be monitored for iron overload that ultimately will require chelation therapy. Table 418,28,29,32,37–45 provides some major indications for transfusion therapy.
TABLE 4 Indications for Episodic and Chronic Blood Transfusions in Patients with Sickle Cell Disease
| Episodic transfusions |
|---|
| Progressive symptoms of acute chest syndrome |
| Multi-organ failure syndrome |
| Acute splenic or hepatic sequestration |
| Major surgical procedures/general anesthesia; tonsillectomy and adenoidectomy should not be considered low-risk procedures.* |
| Acute management of stroke |
| Symptomatic anemia |
| Pregnant patients: medical and obstetric complications |
| Chronic transfusions |
| Primary stroke prevention for at-risk children as identified by transcranial Doppler ultrasonography |
| Prevention of stroke recurrence |
| Chronic heart failure |
| Pulmonary hypertension |
| Prevention of recurrent splenic sequestration |
*—National Institutes of Health guidelines recommend preoperative transfusions for all but lowest risk surgical procedures.
HEMATOPOIETIC CELL TRANSPLANTATION
The only curative treatment for patients with sickle cell disease is allogeneic hematopoietic cell transplantation (HCT).46 There are published reports47 on more than 200 patients with hemoglobin SS disease who were treated with HCT. The disease-free survival rate achieved with HCT ranges from 85 to 90 percent. However, transplant-related mortality is 5 to 10 percent, mainly because of graft-versus-host disease and infection.
Because of even higher mortality in older persons, trials to date typically have included only patients 16 years or younger who have a human leukocyte-antigen–matched sibling and have already had a sickle cell disease–related complication such as stroke, acute chest syndrome, mild chronic lung disease, or red cell alloimmunization resulting from long-term transfusion. Current barriers to the widespread use of transplantation include the lack of human leukocyte-antigen–identical donors and the associated toxicity of current protocols.
Life-Cycle Needs of Patients with Sickle Cell Disease
Family physicians, in collaboration with a sickle cell disease subspecialist, play a critical role in ensuring that patients receive optimal preventive, acute, and chronic care. In addition to the clinical management addressed above, there are specific life-cycle issues that deserve attention. Infants and children clearly benefit from health care supervision provided by a comprehensive sickle cell center. A recent study48 shows that more than 85 percent of children with sickle cell disease who are involved with such a program survive to 18 years of age. The increase in survival of children has led to a burgeoning adult population with sickle cell disease. Young adulthood signals not only the need for greater responsibility in their health care but also potential problems with adjusting to adult-oriented health care settings. The American Academy of Family Physicians has endorsed a consensus statement49 on the need for coordinated health care programs to address these issues during transition to adulthood.
Adult patients require periodic evaluations by a sickle cell disease expert. Additionally, pregnancy represents a time when comanagement with hematology and high-risk obstetric subspecialists can decrease morbidity for mother and child. Because the older patient with sickle cell disease often faces additional problems caused by chronic organ damage, there is a need for subspecialists to be actively involved in ongoing health care. Table 5 provides a list of helpful Internet resources for obtaining updated information on sickle cell disease.
TABLE 5 Internet Resources for Patients with Sickle Cell Disease
| Centers for Disease Control and Prevention Vaccination/Immunization Schedules | |
| Web site:http://www.cdc.gov/nip/recs/child-schedule.htm (children) orhttp://www.cdc.gov/nip/recs/adult-schedule.htm (adults) | |
| Comprehensive Sickle Cell Centers | |
| Web site:http://www.rhofed.com/sickle | |
| Emory University Sickle Cell Information Center | |
| Web site:http://www.scinfo.org/ | |
| NIH Clinical Trials Information | |
| Web site:http://www.clinicaltrials.gov | |
| NIH/NHLBI The Management of Sickle Cell Disease | |
| Web site:http://www.nhlbi.nih.gov/health/prof/blood/sickle/index.htm or http://www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf | |
| Sickle Cell Disease in Children and Adolescents: Diagnosis, Guidelines for Comprehensive Care, and Protocols for Management of Acute and Chronic Complications (Texas Department of State Health Services) | |
| Web site:http://www.dshs.state.tx.us/newborn/sickle.shtm | |
| Sickle Cell Disease Association of America | |
| Web site:http://www.sicklecelldisease.org | |
NIH = National Institutes of Health; NHLBI = National Heart, Lung, and Blood Institute.
