
Am Fam Physician. 2007;75(3):393-394
Author disclosure: Nothing to disclose.
A 26-year-old woman presented with visual changes in her left eye and difficulty walking. She had given birth three months earlier and had noted the enlargement of a scotoma in the temporal visual field of her left eye during pregnancy. She also complained of intermittent paresthesias in her right arm. Her medical history was notable for central nervous system (CNS) hemangioblastomas of the cerebellum and spinal cord at level C7-T1 resected three years earlier. Furthermore, she had previously undergone a partial left nephrectomy for renal cell carcinoma.
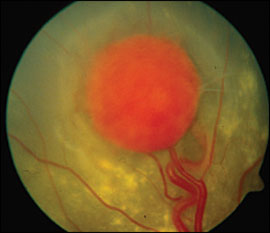
Neurologic examination revealed intact cranial nerves, normal results on strength and sensory examinations, unsteady gait with difficulty performing tandem walk, and a positive result on Romberg's test. On ophthalmic examination, the patient had bilateral 20/30 vision without correction, which was unchanged from three years earlier; the sclera, conjunctiva, cornea, iris, and lens appeared normal. Dilated fundus examination showed a large, exophytic tumor in the left eye. The image of the peripheral fundus demonstrated this large retinal capillary hemangioma, with a dilated feeder arteriole and draining venule (Figure 1). Exudate, seen as yellowish subretinal deposits, had caused localized retinal detachment.
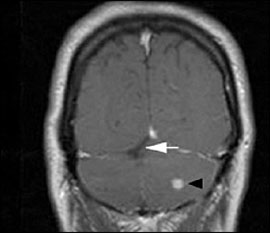
Magnetic resonance imaging of the brain revealed multiple enhancing nodules throughout the cerebellum consistent with hemangioblastomas (Figure 2). Edema was noted bilaterally in the cerebellum, with mild mass effect on the lateral aspect of the fourth ventricle. Postsurgical changes also were noted in the cerebellum.
Question
Discussion
The correct answer is D: von Hippel-Lindau disease. In 1904, von Hippel, a German ophthalmologist, described two patients with retinal angiomatosis,1 and in 1926, Lindau, a Swedish neurologist, described an association among retinal angiomatosis, hemangiomatous cysts of the cerebellum, and the visceral components of the disease.2 The transmission of von Hippel-Lindau disease is as an autosomal dominant trait with irregular penetrance.3
Cerebellar hemangioblastoma is the characteristic CNS lesion. In a study of 152 patients with von Hippel-Lindau disease, cerebellar hemangioblastomas were present in 60 percent of the patients.4 Hemangioblastomas also may occur in the medulla oblongata and spinal cord. Progressive enlargement of tumors associated with von Hippel-Lindau disease during pregnancy has been described for CNS lesions5 and retinal lesions.6 Renal cell carcinoma occurs in approximately one fourth of affected patients.7 The main causes of death are metastatic renal cell carcinoma and complications from CNS hemangioblastomas.
In published case series, pheochromocytoma occurs in 3 to 10 percent of patients, with a tendency to cluster in families.8 Other visceral manifestations include renal cysts, pancreatic cysts, neuroendocrine tumors, adenomas of the epididymis and the kidneys, endolymphatic sac tumors of the inner ear, and hepatic and epididymal cysts. Typically asymptomatic, these lesions may help diagnose von Hippel-Lindau disease.9
The ophthalmic manifestations are often among the first to be diagnosed. Retinal capillary hemangiomas may develop in 40 to 60 percent of patients with von Hippel-Lindau disease. Although the hemangiomas may be present at birth, they typically are not detected until the second or third decade of life because of their small size and peripheral location.10 Treatment depends on the location and size of the lesions. Small lesions and those touching the optic nerve are more likely to be observed.11 Larger lesions may be treated with laser photocoagulation, cryotherapy, or photodynamic therapy. The visual prognosis for patients with retinal capillary hemangioma is guarded, especially with large, untreated lesions.
Patients with ataxia-telangiectasia may have multiple telangiectatic vascular malformations on the skin and conjunctiva, but generally these malformations are absent from the retina and the CNS. The hallmark of the disorder is cerebellar cortical atrophy as well as immunodeficiency.
Sturge-Weber syndrome is characterized by a facial port-wine stain, glaucoma, and diffuse choroidal hemangioma, which gives the fundus a “tomato ketchup” appearance. They also may have ipsilateral leptomeningeal hemangiomas.
Tuberous sclerosis is characterized by an adenoma sebaceum rash on the nose, cheeks, and forehead and is associated with infantile spasms and mental retardation. Patients with tuberous sclerosis also may have astrocytic hamartomas, which are small, white masses in the retina and near the optic nerve.
The clinical appearance of Wyburn-Mason's syndrome is different, with direct arteriovenous communications between retinal arteries and veins but no intervening capillary bed. Affected patients may have intracranial arteriovenous malformations.
| Condition | Characteristics |
|---|---|
| Ataxia-telangiectasia | Telangiectasia of skin and conjunctiva, cerebellar ataxia |
| Sturge-Weber syndrome | Congenital facial angioma, glaucoma, choroidal hemangioma, leptomeningeal hemangioma |
| Tuberous sclerosis | Astrocytic retinal hamartoma, facial angiofibroma, infantile spasms |
| von Hippel-Lindau disease | Retinal capillary hemangioma, associated with cerebellar vascular tumors, renal cell carcinoma, and pheochromocytoma |
| Wyburn-Mason's syndrome | Retinal arteriovenous communications, associated with intracranial arteriovenous malformations |