Turner syndrome occurs in one out of every 2,500 to 3,000 live female births. The syndrome is characterized by the partial or complete absence of one X chromosome (45,X karyotype). Patients with Turner syndrome are at risk of congenital heart defects (e.g., coarctation of aorta, bicuspid aortic valve) and may have progressive aortic root dilatation or dissection. These patients also are at risk of congenital lymphedema, renal malformation, sensorineural hearing loss, osteoporosis, obesity, diabetes, and atherogenic lipid profile. Patients usually have normal intelligence but may have problems with nonverbal, social, and psychomotor skills. Physical manifestations may be subtle but can include misshapen ears, a webbed neck, a broad chest with widely spaced nipples, and cubitus valgus. A Turner syndrome diagnosis should be considered in girls with short stature or primary amenorrhea. Patients are treated for short stature in early childhood with growth hormone therapy, and supplemental estrogen is initiated by adolescence for pubertal development and prevention of osteoporosis. Almost all women with Turner syndrome are infertile, although some conceive with assisted reproduction.
Turner syndrome is diagnosed in females with partial or complete absence of one X chromosome (45,X karyotype). Clinical manifestations vary and may be subtle, but they usually include short stature, a broad chest with widely spaced nipples, cubitus valgus, congenital lymphedema, and a lack of spontaneous pubertal development from ovarian sex hormone insufficiency.1
Turner syndrome occurs in one out of 2,500 to 3,000 live female births2; however, many more 45,X conceptuses do not survive past the first trimester. Ninety-nine percent of conceptuses with a 45,X karyotype abort spontaneously; Turner syndrome causes 10 percent of all first-trimester miscarriages.3 Unlike with Down syndrome, maternal age does not increase the risk of Turner syndrome, and there are no clearly established risk factors. Recurrence in subsequent pregnancies is rare.4
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| If any Y chromosome material is shown on the karyotype, prophylactic laparoscopic gonadectomy is required. | C | 11 |
| Cardiovascular evaluations should be performed at diagnosis to rule out congenital heart defects. | C | 11 |
| Ongoing estrogen therapy should be initiated in the preteen years. | B | 17 |
| Short stature should be treated with human growth hormone until the patient reaches a bone age of 14 years. | A | 15,16 |
| Calcium and vitamin D supplementation should be initiated at 10 years of age. | C | 11 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, see page 323 or https://www.aafp.org/afpsort.xml.
Etiology
Turner syndrome is caused by a reduced complement of genes that are typically expressed from both X chromosomes in females. Normally one X chromosome is randomly inactivated during the first week of life (when there are fewer than 200 embryonic cells); therefore, it may seem paradoxical that having a single X chromosome would cause clinical consequences. However, not all genes from the second chromosome are inactivated in Turner syndrome. Some genes escape X-inactivation via a process initiated by the X-inactivation-specific transcript (XIST) gene that is transcribed exclusively from the inactive genes. The loss of these noninactivated X genes causes the phenotypic manifestations characteristic of Turner syndrome, such as short stature.5
Clinical Presentation
The presentation of Turner syndrome varies throughout a patient's life. The diagnosis should be considered in a female fetus with hydrops, increased nuchal translucency, cystic hygroma, or lymphedema.6 At any age, Turner syndrome may be difficult to recognize clinically because the characteristic facial features can be subtle (Figure 1). Key clinical features of Turner syndrome are a lack of breast development or amenorrhea, with elevated follicle-stimulating hormone levels by 14 years of age; and infertility in women. Other characteristics of Turner syndrome include short stature, a webbed neck, a low posterior hairline, misshapen or rotated ears, a narrow palate with crowded teeth, a broad chest with widely spaced nipples, cubitus valgus, hyperconvex nails, multipigmented nevi, pubertal delay, and cardiac malformation.1
Figure 1.
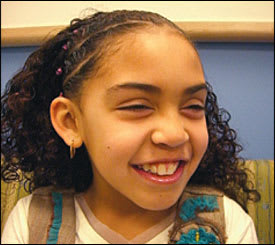
A 12-year-old girl with Turner syndrome. Note the subtle, distinctive facial features including prominent, posteriorly rotated auricles with looped helices and attenuated tragi; infraorbital skin creases; and mildly foreshortened mandible.
One third of patients with Turner syndrome have a cardiac malformation; 75 percent of these patients have coarctation of aorta or a bicuspid aortic valve.7 Progressive aortic root dilatation or dissection can also occur, particularly in patients with a bicuspid valve, coarctation, or untreated hypertension.8,9 Patients with Turner syndrome often have an atherogenic cardiovascular risk factor profile.10 Other potential complications of Turner syndrome include strabismus, sensorineural hearing loss, recurrent otitis media, orthodontic anomalies, renal malformation (e.g., horseshoe kidney, duplicated or cleft renal pelvis), autoimmune thyroiditis, celiac disease, congenital hip dysplasia, and scoliosis.11
Girls with Turner syndrome typically have normal intelligence (i.e., mean full scale IQ of 90); however, they may have difficulty with nonverbal, social, and psychomotor skills. If development is frankly delayed, an alternative explanation should be considered, along with prompt referral for early intervention.12
Diagnosis
A diagnosis of Turner syndrome can be confirmed with standard karyotyping (i.e., chromosomal analysis of 30 peripheral lymphocytes). More than one half of patients with the condition will have a missing X chromosome (45,X) in all cells studied or a combination of monosomy X and normal cells (45,X/46,XX; mosaic Turner syndrome). A mosaic result does not necessarily predict severity because karyotyping only investigates lymphocytes, not the relevant tissues (e.g., brain, heart, ovaries).
A karyotype is obtained by sending whole blood, at room temperature and in a green-top sodium heparin tube, to a laboratory for testing. Karyotyping takes about one week. If an urgent result is needed (e.g., because of parental anxiety or a critical clinical situation), X-specific fluorescence in situ hybridization can confirm monosomy X in less than 24 hours.
Although 45,X is the karyotype typically seen in patients with Turner syndrome, other sex chromosome anomalies such as isochromosome Xq, ring X, deletion Xp, or an abnormal Y chromosome can also cause the condition.4 Patients with Y chromosome material have a 12 percent risk of gonadoblastoma and must be referred for imaging studies and laparoscopic removal of testicular tissue (i.e., gonadectomy).11
Delayed diagnosis of Turner syndrome in girls with short stature is typical. One study showed that the diagnosis is made an average of seven years after short stature is clinically evident on female growth curves.13 In a case series, 4 percent of girls referred for genetic evaluation of isolated short stature, regardless of familial background height, were diagnosed with Turner syndrome.14 More than 30 percent of the referred girls who had amenorrhea or suggestive phenotypic features had Turner syndrome.14 Karyotyping is indicated for girls with unexplained short stature (more than two standard deviations below the mean height for age).13,14
Management
Given the complexity and multisystem nature of Turner syndrome, family physicians can play an important role in coordinating multidisciplinary management and in directly managing risk factors and complications (e.g., infertility, cardiovascular complications, osteoporosis). Controlled studies with patient-oriented outcomes such as morbidity, mortality, and quality of life in patients with Turner syndrome are generally lacking. However, the Turner Syndrome Consensus Study Group, sponsored by the National Institutes of Health's National Institute of Child Health and Human Development, has published comprehensive management guidelines based on collective expert opinion and a review of existing literature (Table 1).11,15–17
Table 1 Guidelines for the Management of Turner Syndrome
| System | Timing | Clinical issue | Intervention | Evidence level |
|---|---|---|---|---|
| Auditory | At diagnosis and every one to five years thereafter | Sensorineural hearing loss | Hearing evaluation; audiology; hearing aids | C11 |
| Childhood | Recurrent otitis media | Pressure-equalizing tubes for middle ear effusion in patients older than three months | C11 | |
| Bones | From 10 years of age to adulthood | Osteopenia; osteoporosis | Elemental calcium (1,200 to 1,500 mg per day); vitamin D supplementation; appropriate estrogen therapy; exercise | C11 |
| First adult office visit | Bone mineral density | Baseline dual energy x-ray absorptiometry scan | C11 | |
| Mid- to late adulthood | Osteoporosis | Bisphosphonate therapy (if high risk) | C11 | |
| Cardiovascular | At diagnosis | Congenital heart defects | Cardiovascular evaluation; echocardiography or MRI; ECG | C11 |
| Every five to 10 years (adulthood) | Aortic root dilatation | Echocardiography or MRI | C11 | |
| All ages | Hypertension | Blood pressure in all four extremities | C11 | |
| Older girls/adulthood | Hyperlipidemia | Annual fasting lipid screening | C11 | |
| Dental | Seven years and older | Malocclusion and other tooth anomalies | Orthodontic evaluation | C11 |
| Genetics | All ages | Presence of Y chromosome material | Laparoscopic gonadectomy to prevent gonadoblastoma | C11 |
| Immune | At diagnosis and annually thereafter | Thyroiditis (hypo- or hyperthyroid) | Thyroid function tests (i.e., thyroxine and thyroid-stimulating hormone levels) | C11 |
| Every two to four years after four years of age | Celiac disease | Tissue transglutaminase immunoglobulin A measurement | C11 | |
| Hepatic | Every one to two years after six years of age | Liver enzymes persistently elevated for more than six months | Ultrasonography to evaluate for hepatic steatosis; hepatology consult | C11 |
| Lymphatic | Usually younger than two years of age | Lymphedema | Support stockings; decongestive physiotherapy | C11 |
| Metabolic | Older girls/adulthood | Diabetes | Annual fasting plasma glucose screening | C11 |
| All ages | Obesity | Target body mass index less than 25 kg per m2 | C11 | |
| Skeletal/growth | Nine to 24 months of age to adulthood (bone age of 14 years) | Short stature (i.e., more than two standard deviations below the mean) | Human growth hormone with or without oxandrolone (Oxandrin) | A15,16 |
| Infancy to four years of age | Hip dislocation | Physical examination with Barlow/Ortolani maneuvers | C11 | |
| Teenagers | Scoliosis; kyphosis | Physical examination with a scoliometer | C11 | |
| Psychological | All ages | Self-esteem; learning issues | Psychoeducational evaluation (school based); support | C11 |
| Ophthalmologic | At diagnosis if older than one year | Strabismus; hyperopia | Ophthalmologic evaluation | C11 |
| Renal | At diagnosis | Congenital renal malformations | Renal ultrasonography | C11 |
| Reproductive | Preteen | Puberty | Estrogen therapy | B17 |
| Adulthood | Planned pregnancy | Echocardiography or cardiac MRI; high-risk consult | C11 | |
| Adulthood | Infertility | Assisted reproduction or infertility consult | C11 | |
| Adulthood | Estrogen deficiency | Female sex hormone replacement | B17 |
MRI = magnetic resonance imaging; ECG = electrocardiography.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series.
CHILDREN
The key aspects of managing Turner syndrome in children are cardiovascular monitoring and treatment of congenital heart disease; growth hormone therapy to augment linear growth (as early as 12 to 24 months of age); and supplemental estrogen therapy for sexual development and preservation of bone mineral density (typically initiated in the preteen years).17 The mean adult height in patients with Turner syndrome is 4 ft, 8 in (140 cm); but with growth hormone and estrogen therapy, the average height increases to 5 ft (150 cm).15,16,18 Growth hormone therapy is typically discontinued after the patient reaches a bone age of 14 years; sex hormone therapy is generally continued throughout life.15,16
Patients with Turner syndrome require audiometry at diagnosis and periodically thereafter to assess for sensorineural or conductive hearing loss from recurrent otitis media; blood pressure measurement in all four extremities; and ongoing annual thyroid function, liver enzyme, and fasting lipid and glucose monitoring. Infants and young children with Turner syndrome should be examined with Barlow/Ortolani maneuvers for evidence of congenital hip dislocation, and those older than one year should be referred to a pediatric ophthalmologist to assess for hyperopia and strabismus. Ultrasonography should be performed at diagnosis to assess for congenital renal malformations. Girls older than four years should have a tissue transglutaminase immunoglobulin A measurement every two to four years to detect celiac disease. Patients seven years or older need orthodontic evaluation for malocclusion or other tooth anomalies. Teenagers should be carefully monitored for scoliosis and kyphosis.
ADULTS
Fertility and sexual development are often major concerns for patients with Turner syndrome. The ovaries develop but typically degenerate during fetal life or in early childhood. However, spontaneous menstruation and childbirth occur in 2 to 5 percent of patient with Turner syndrome, which may be explained by substantial 46,XX/45,X mosaicism, with normal cell populations existing in the ovaries.19 Because spontaneous pregnancy is possible, patients should be counseled about birth control if sexually active.
Patients with Turner syndrome are likely to ask their family physicians about reproductive potential, and age-appropriate counseling about infertility treatments can markedly mitigate the adverse psychological impact of the diagnosis.12 In vitro fertilization (using oocytes harvested and cryopreserved before ovarian regression is complete) is being studied in young women with Turner syndrome.11 Spontaneous or assisted pregnancy carries substantial risks; therefore, preconception counseling and cardiac echocardiography or magnetic resonance imaging (MRI) are essential. Primary care providers should monitor the pregnancy as part of a multidisciplinary team (e.g., high-risk obstetrics, cardiology, and reproductive endocrinology subspecialists).
In addition to reproductive counseling, the transition to adult treatment of Turner syndrome includes management of atherogenic cardiovascular risk factors (e.g., hypertension, diabetes, hyperlipidemia); calcium and vitamin D supplementation to prevent osteoporosis; and ongoing sex hormone therapy. A baseline dual energy x-ray absorptiometry scan to evaluate bone mineral density is recommended at the first adult visit. Adult women should continue to receive high-quality echocardiography or MRI of the aorta every five to 10 years to assess the need for surgical correction of severe aortic root dilatation (which occurs over time in 8 to 42 percent of patients).
PSYCHOSOCIAL EFFECTS
The psychosocial impact of Turner syndrome may be substantial for young girls and women. These effects may be caused by (in decreasing order of patient importance) infertility; short stature; and impaired development of sexual characteristics, most importantly lack of libido.12 Physicians should elicit specific concerns from patients, addressing them individually, and should recommend comprehensive school-based psychoeducational assessment.
