Pulmonary arterial hypertension is defined as a mean pulmonary arterial pressure greater than 25 mm Hg at rest or 30 mm Hg during physical activity. Pulmonary arterial hypertension is classified into subgroups, including idiopathic, heritable, and pulmonary arterial hypertension associated with other conditions. A detailed history, thorough physical examination, and most importantly, a high index of suspicion are essential to diagnosis. Evaluation includes echocardiography and exclusion of other causes of symptoms. Targeted laboratory testing can help identify the subgroup of pulmonary arterial hypertension. Right heart catheterization is required to confirm the diagnosis. Standard treatment options include oral anticoagulation, diuretics, oxygen supplementation, and for a small percentage of patients, calcium channel blockers. Newer treatments include prostacyclin analogues, endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors. Combination therapy has been shown to improve pulmonary arterial pressure, but more research is needed. Interventional procedures for patients with pulmonary arterial hypertension include balloon atrial septostomy and lung transplantation.
Pulmonary arterial hypertension (PAH) is a rare, underdiagnosed condition defined as elevation of mean pulmonary arterial pressure. In patients with PAH, the average pulmonary arterial pressure is greater than 25 mm Hg at rest (compared with 15 mm Hg in patients without PAH) or 30 mm Hg during physical activity, as measured by right heart catheterization. The prevalence of PAH varies among specific populations, but one study estimated that it affects 15 in 1 million adults.1 Idiopathic PAH occurs mainly in persons in their 20s and 30s, with an overall female-to-male ratio of 1.7:1, which is higher in black persons (4.3:1).2 Risk factors for PAH include a family history of PAH, congenital heart disease, connective tissue disease, portal hypertension, sickle cell disease, thyroid disease, human immunodeficiency virus (HIV), and use of certain drugs and toxins. Table 1 lists the clinical classification of PAH, which was updated in 2008.3
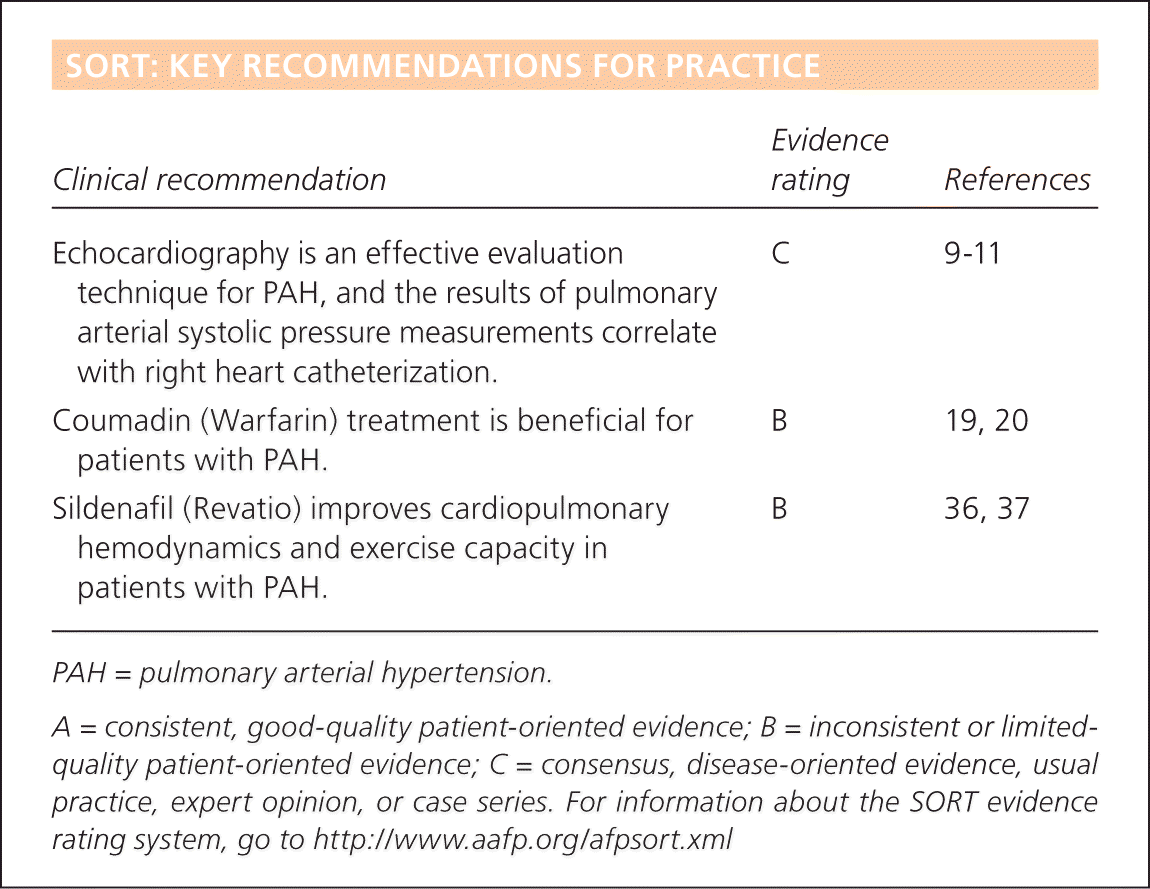
SORT: KEY RECOMMENDATIONS FOR PRACTICE
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Echocardiography is an effective evaluation technique for PAH, and the results of pulmonary arterial systolic pressure measurements correlate with right heart catheterization. | C | 9–11 |
| Coumadin (Warfarin) treatment is beneficial for patients with PAH. | B | 19, 20 |
| Sildenafil (Revatio) improves cardiopulmonary hemodynamics and exercise capacity in patients with PAH. | B | 36, 37 |
PAH = pulmonary arterial hypertension.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidencerating system, go to https://www.aafp.org/afpsort.xml
Table 1. Clinical Classification of PAH

| Idiopathic | ||
| Heritable | ||
| BMPR2 gene | ||
| ALK1 gene, endoglin (with or without hereditary hemorrhagic telangiectasia) | ||
| Unknown | ||
| Drug- and toxin-induced | ||
| Definite causal relationship | ||
| Aminorex* | ||
| Dexfenfluramine* | ||
| Fenfluramine* | ||
| Toxic rapeseed oil | ||
| Likely causal relationship | ||
| Amphetamines | ||
| L-tryptophan | ||
| Methamphetamines | ||
| Possible causal relationship | ||
| Chemotherapeutic agents | ||
| Cocaine | ||
| Phenylpropanolamine* | ||
| Selective serotonin reuptake inhibitors | ||
| St. John's wort | ||
| Unlikely causal relationship | ||
| Cigarette smoking | ||
| Estrogen | ||
| Oral contraceptives | ||
| Conditions associated with PAH | ||
| Chronic hemolytic anemia | ||
| Congenital heart disease | ||
| Connective tissue disease | ||
| Human immunodeficiency virus infection | ||
| Portal hypertension | ||
| Schistosomiasis | ||
| Persistent pulmonary hypertension of the newborn | ||
PAH = pulmonary arterial hypertension.
*—Not available in the United States.
Information from reference 3.
Clinical Features
The early phases of PAH may be asymptomatic. Patients usually present with dyspnea exacerbated by exertion, fatigue, chest pain, and palpitations. The diagnosis of PAH is often delayed because many other conditions can cause similar symptoms. The differential diagnosis includes congestive heart failure, coronary artery disease, pulmonary embolism, and chronic obstructive pulmonary disease. Advanced PAH may present as clinically evident right-sided heart failure, dizziness, syncope, edema, or cyanosis.
If PAH is incidentally discovered, the physician should attempt to find or exclude possible modifiable causes. Confirmation by right heart catheterization should be considered before beginning an extensive search for an underlying cause, starting therapy, or providing prognostic advice.4
Diagnosis
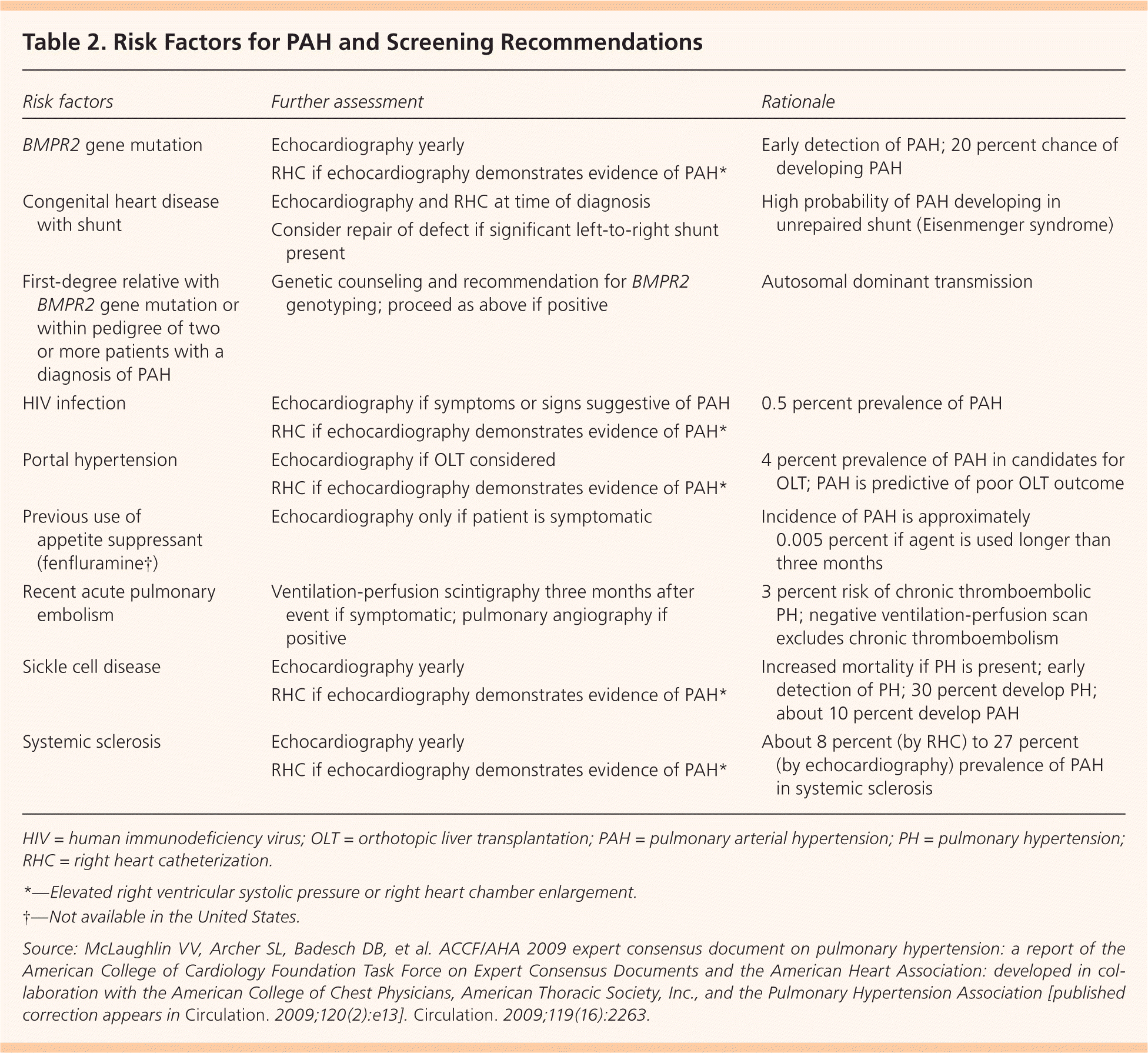
A detailed history, thorough physical examination, and most importantly, a high index of suspicion are essential to diagnosing PAH.5 A French study reported an average delay of 27 months from the onset of symptoms to diagnosis.1 PAH should be considered in any patient with dyspnea who shows no or minimal signs of specific heart or lung disease. Suspicion should be especially high in patients who also have conditions associated with PAH, such as sickle cell anemia, HIV, and systemic sclerosis. The American College of Cardiology Foundation and the American Heart Association offer screening guidelines for patients with known risk factors for PAH (Table 2).6
Table 2. Risk Factors for PAH and Screening Recommendations
| Risk factors | Further assessment | Rationale |
|---|---|---|
| BMPR2 gene mutation | Echocardiography yearly | Early detection of PAH; 20 percent chance of developing PAH |
| RHC if echocardiography demonstrates evidence of PAH* | ||
| Congenital heart disease with shunt | Echocardiography and RHC at time of diagnosis | High probability of PAH developing in unrepaired shunt (Eisenmenger syndrome) |
| Consider repair of defect if significant left-to-right shunt present | ||
| First-degree relative with BMPR2 gene mutation or within pedigree of two or more patients with a diagnosis of PAH | Genetic counseling and recommendation for BMPR2 genotyping; proceed as above if positive | Autosomal dominant transmission |
| HIV infection | Echocardiography if symptoms or signs suggestive of PAH | 0.5 percent prevalence of PAH |
| RHC if echocardiography demonstrates evidence of PAH* | ||
| Portal hypertension | Echocardiography if OLT considered | 4 percent prevalence of PAH in candidates for OLT; PAH is predictive of poor OLT outcome |
| RHC if echocardiography demonstrates evidence of PAH* | ||
| Previous use of appetite suppressant (fenfluramine†) | Echocardiography only if patient is symptomatic | Incidence of PAH is approximately0.005 percent if agent is used longer than three months |
| Recent acute pulmonary embolism | Ventilation-perfusion scintigraphy three months after event if symptomatic; pulmonary angiography if positive | 3 percent risk of chronic thromboembolic PH; negative ventilation-perfusion scan excludes chronic thromboembolism |
| Sickle cell disease | Echocardiography yearly | Increased mortality if PH is present; early detection of PH; 30 percent develop PH; about 10 percent develop PAH |
| RHC if echocardiography demonstrates evidence of PAH* | ||
| Systemic sclerosis | Echocardiography yearly | About 8 percent (by RHC) to 27 percent(by echocardiography) prevalence of PAH in systemic sclerosis |
| RHC if echocardiography demonstrates evidence of PAH* |
HIV = human immunodeficiency virus; OLT = orthotopic liver transplantation; PAH = pulmonary arterial hypertension; PH = pulmonary hypertension; RHC = right heart catheterization.
*—Elevated right ventricular systolic pressure or right heart chamber enlargement.
†—Not available in the United States.
Source: McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association [published correction appears in Circulation. 2009;120(2):e13]. Circulation. 2009;119(16):2263.
Physical examination of a patient with PAH may reveal a right parasternal lift, accentuated pulmonary second heart sound, pansystolic murmur (tricuspid regurgitation), third heart sound, and a diastolic murmur (pulmonary valve insufficiency). Edema may present as jugular vein distension, hepatomegaly, peripheral edema, or ascites.6
DIAGNOSTIC TESTING
Diagnostic testing options for PAH are listed in Table 3.6 Electrocardiography (ECG) may show right ventricular hypertrophy and right axis deviation, but ECG has inadequate sensitivity and specificity to reliably detect PAH.7 One study showed that a combination of P- and T-wave indicators may provide an effective means of assessing treatment response in patients with PAH.8
Table 3. Diagnostic Testing Options for PAH
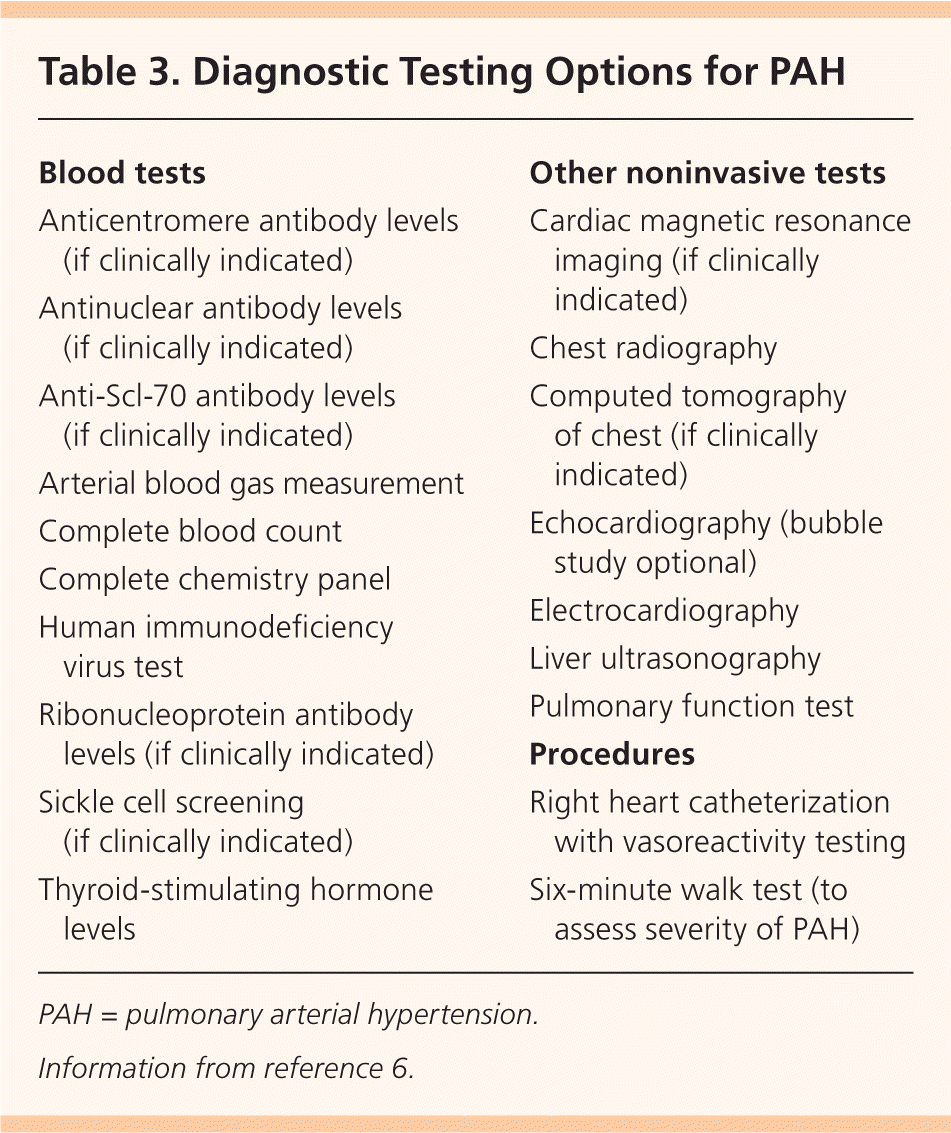
| Blood tests |
| Anticentromere antibody levels (if clinically indicated) |
| Antinuclear antibody levels (if clinically indicated) |
| Anti-Scl-70 antibody levels (if clinically indicated) |
| Arterial blood gas measurement |
| Complete blood count |
| Complete chemistry panel |
| Human immunodeficiency virus test |
| Ribonucleoprotein antibody levels (if clinically indicated) |
| Sickle cell screening (if clinically indicated) |
| Thyroid-stimulating hormone levels |
| Other noninvasive tests |
| Cardiac magnetic resonance imaging (if clinically indicated) |
| Chest radiography |
| Computed tomography of chest (if clinically indicated) |
| Echocardiography (bubble study optional) |
| Electrocardiography |
| Liver ultrasonography |
| Pulmonary function test |
| Procedures |
| Right heart catheterization with vasoreactivity testing |
| Six-minute walk test (to assess severity of PAH) |
PAH = pulmonary arterial hypertension.
Information from reference 6.
Chest radiography is abnormal in 90 percent of patients at diagnosis and usually shows right ventricular enlargement, a prominent central pulmonary artery, or peripheral hypovascularity7 (Figure 1). A normal chest radiograph does not rule out the diagnosis.7
Figure 1.
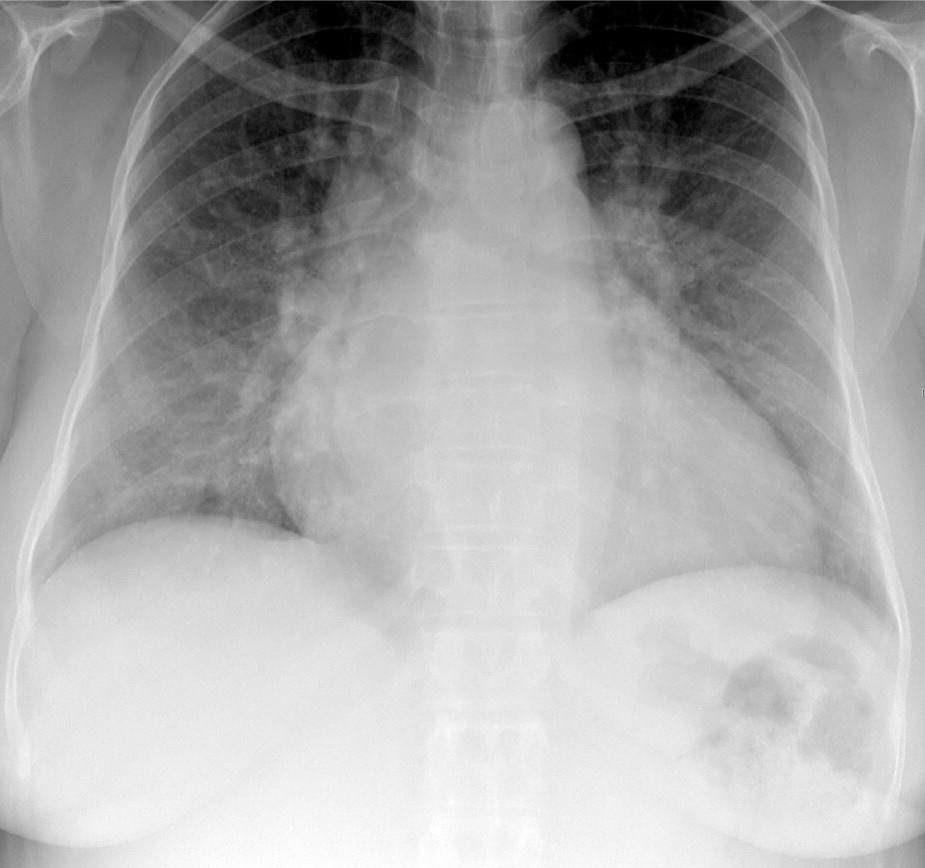
Chest radiography shows bilateral hilar enlargement, which is a typical finding in patients with pulmonary arterial hypertension, as well as cardiomegaly.
Studies have shown a correlation in pulmonary arterial systolic pressure measurement between transthoracic echocardiography and right heart catheterization.9–11 Echocardiography has been used to identify PAH in patients with associated conditions.12–14 Measuring parameters such as septal systolic strain and strain rate may allow for earlier detection of PAH.15 Increased tricuspid regurgitation, and right ventricular and atrial enlargement are echocardiographic signs of PAH.7
PAH must be distinguished from other causes of pulmonary hypertension. Pulmonary function tests and arterial blood gas measurements help identify underlying lung disease. Patients may have mild to moderate reduction of lung volumes on these tests, but the partial arterial oxygen tension (PaO2) is normal or only slightly decreased and the partial arterial carbon dioxide tension (PaCO2) is usually decreased from hyperventilation.7 High-resolution computed tomography can assist in the evaluation of thromboembolic disease, emphysema, or interstitial lung disease. Although cardiac magnetic resonance imaging (MRI) is not currently used as a routine assessment tool, continued improvement in MRI acquisition and spatial resolution may lead to improved understanding of PAH pathophysiology, and aid in diagnosis and prognosis. Cardiac MRI may replace more invasive diagnostic tests.16
Evidence of associated conditions may identify the type of PAH. Routine chemistry panels, complete blood count, and thyroid function tests should be obtained. Antinuclear antibody (ANA), anti-Scl-70, anticentromere, and ribonucleoprotein antibody levels are recommended to evaluate for connective tissue disease.7 About one third of patients with idiopathic PAH have low ANA titers (less than 1:80). If the ANA levels are elevated or if clinical features suggestive of connective tissue disease are present, rheumatologic consultation is recommended.7 All patients should be tested for HIV.7 Liver ultrasonography is a reliable test to exclude cirrhosis or portal hypertension.7
CONFIRMATORY AND PROGNOSTIC TESTING
Right heart catheterization is required to confirm the diagnosis of PAH and to evaluate the severity of hemo-dynamic dysfunction. This should be performed at an institution with considerable experience with PAH, specifically with vasoreactivity testing because of potential procedural complications.6 Vasoreactivity testing is also needed to identify patients who may benefit from treatment with calcium channel blockers.17 Patients with a mean pulmonary arterial pressure decrease of more than 10 mm Hg to below 40 mm Hg and with an unchanged or increased cardiac output when challenged are considered vasoreactive.6 Lung biopsy is not recommended because it has considerable risks and a very low likelihood of altering the diagnosis or treatment.6
The best test to classify the severity of PAH and estimate prognosis is the six-minute walk test. This simple test measures the distance walked in six minutes. It is predictive of survival in idiopathic PAH.18 Research is ongoing to better assess prognosis for patients with all subtypes of PAH.6
Treatment
Because of the complexity of treatment, patients should be followed regularly at an institution that specializes in PAH.6 Family physicians should be aware of PAH treatments and their potential adverse effects. Figure 2 presents an algorithm for the treatment of PAH.6
Figure 2. Treatment of PAH
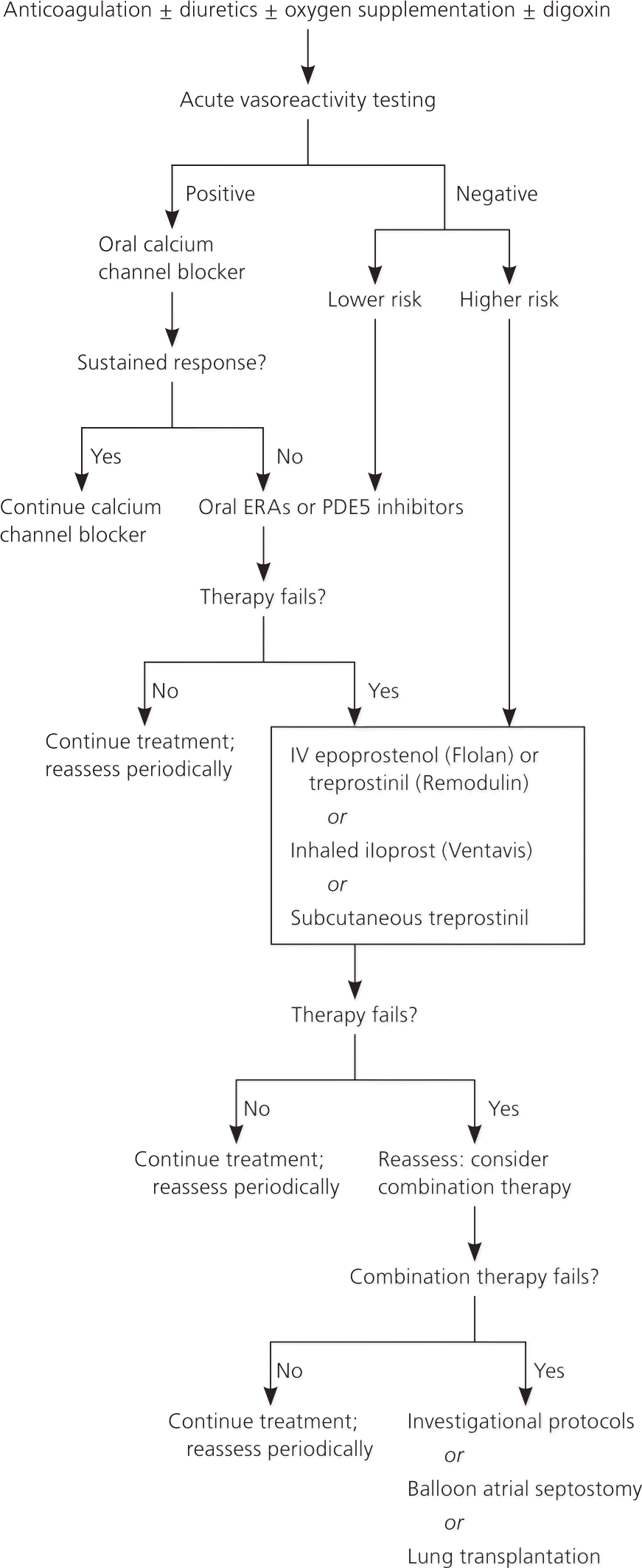
Algorithm for the treatment of PAH. (ERAs = endothelin receptor antagonists; IV = intravenous; PAH = pulmonary arterial hypertension; PDE5 = phosphodiesterase type 5.)
Source: McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association [published correction appears in Circulation . 2009;120(2):e13]. Circulation. 2009;119(16):2276.
STANDARD TREATMENTS
A number of established treatment options for PAH continue to be useful. Two retrospective analyses have reported favorable results with the oral anticoagulant warfarin (Coumadin).19,20 Although there is no expert consensus, an International Normalized Ratio of 1.5 to 2.5 is recommended.6 Patients with right-sided heart failure may benefit symptomatically from diuretics, although no randomized controlled trials have been performed to demonstrate improvement in symptoms or outcomes with diuretic use.6
Most patients with PAH have only mild arterial hypoxia at rest. Although symptomatic improvement has been reported with oxygen supplementation, this has not been confirmed in controlled studies.6 An oxygen saturation greater than 90 percent at all times is recommended.6 Inotropic agents, such as digoxin, have been considered for treatment, but no data are available on the effects of long-term treatment.6
Less than 10 percent of patients with idiopathic PAH have a clinically significant reduction of pulmonary arterial pressure with calcium channel blocker therapy.17
NEWER TREATMENTS
More recent medications for PAH include prostacyclin analogues, endothelin receptor antagonists (ERAs), and phosphodiesterase type 5 inhibitors (Table 46,21–38). Prostacyclin is a potent vasodilator that maintains low vascular tone, has antiproliferative properties, and prevents platelet aggregation. The prostacyclin analogues currently approved in the United States are epoprostenol (Flolan), iloprost (Ventavis), and treprostinil (Remodulin).
Table 4. New Medications for PAH
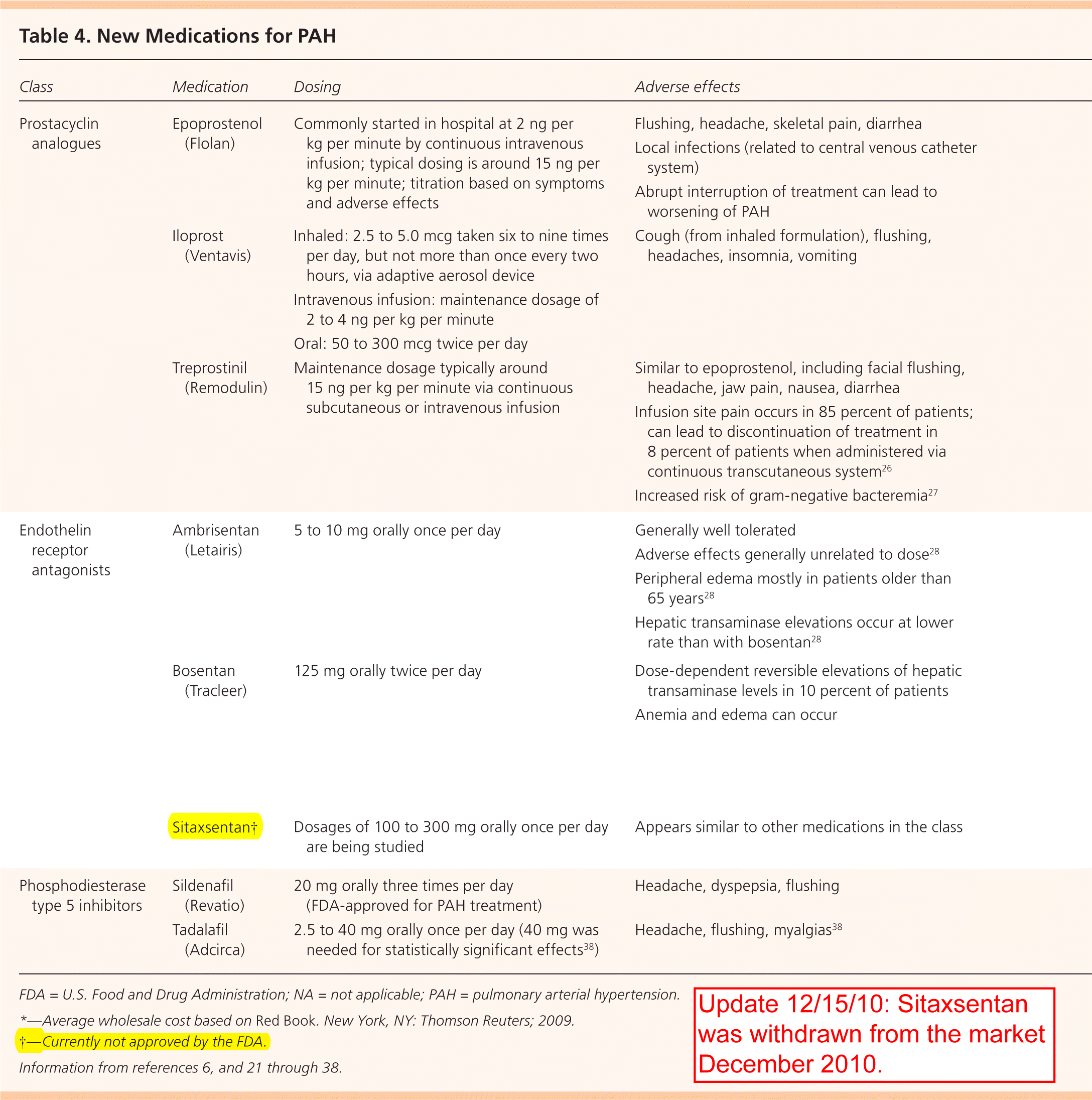
| Class | Medication | Dosing | Adverse effects | Comments | Cost per unit* | Cost per day |
|---|---|---|---|---|---|---|
| Prostacyclin analogues | Epoprostenol (Flolan) | Commonly started in hospital at 2 ng per kg per minute by continuous intravenous infusion; typical dosing is around 15 ng per kg per minute; titration based on symptoms and adverse effects | Flushing, headache, skeletal pain, diarrhea Local infections (related to central venous catheter system) Abrupt interruption of treatment can lead to worsening of PAH | First medication shown to improve survival in idiopathic PAH21 Improves symptoms, exercise capacity, and hemodynamic measures22,23 Recommended for unstable patients in functional classes III and IV 6 | $52 for 1.5-mg intravenous dose | $52, based on 15 ng per kg per minute for patient weighing 70 kg (154 lb) |
| Iloprost (Ventavis) | Inhaled: 2.5 to 5.0 mcg taken six to nine times per day, but not more than once every two hours, via adaptive aerosol device Intravenous infusion: maintenance dosage of 2 to 4 ng per kg per minute Oral: 50 to 300 mcg twice per day | Cough (from inhaled formulation), flushing, headaches, insomnia, vomiting | Inhaled formulation shows small but statistically significant improvement of symptoms in patients with severe PAH24 Approved for patients in functional classes III and IV | $39 for 2-mL inhalation (10 mcg per mL) | $39, based on 2.5 mcg via inhaled formulation eight times per day | |
| Treprostinil (Remodulin) | Maintenance dosage typically around 15 ng per kg per minute via continuous subcutaneous or intravenous infusion | Similar to epoprostenol, including facial flushing, headache, jaw pain, nausea, diarrhea Infusion site pain occurs in 85 percent of patients; can lead to discontinuation of treatment in 8 percent of patients when administered via continuous transcutaneous system26 Increased risk of gram-negative bacteremia27 | Improves exercise capacity; hemodynamics and survival similar to epoprostenol25 Approved for patients in functional classes II and III; intravenous use approved in patients who cannot tolerate subcutaneous infusion6 | $1,345 for 20-mL multidose vial (1 mg per mL) | $100, based on 15 ng per kg per minute for patient weighing 70 kg | |
| Endothelin receptor antagonists | Ambrisentan (Letairis) | 5 to 10 mg orally once per day | Generally well tolerated Adverse effects generally unrelated to dose28 Peripheral edema mostly in patients older than 65 years28 Hepatic transaminase elevations occur at lower rate than with bosentan28 | Prescribed only through the Letairis Education and Access Program Specific to endothelin A receptor Approved in the United States in 2007 for patients in functional classes II and III29,30 FDA requires liver function testing once per month and hematocrit testing every three months Contraindicated in pregnancy | $192 for 10-mg tablet | $192 for 10 mg per day |
| Bosentan (Tracleer) | 125 mg orally twice per day | Dose-dependent reversible elevations of hepatic transaminase levels in 10 percent of patients Anemia and edema can occur | Nonspecific endothelin receptor antagonist Improves hemodynamics, exercise capacity, and echocardiographic results; delays time to clinical worsening31–33 Approved for patients with symptomatic heart failure; benefits shown in patients in functional class II34 FDA requires liver function testing once per month and hematocrit testing every three months | $96 for 125-mg tablet | $192 | |
| Sitaxsentan† | Dosages of 100 to 300 mg orally once per day are being studied | Appears similar to other medications in the class | Specific to endothelin A receptor Benefit and safety demonstrated,35 although currently not approved by the FDA | NA | NA | |
| Phosphodiesterase type 5 inhibitors | Sildenafil (Revatio) | 20 mg orally three times per day (FDA-approved for PAH treatment) | Headache, dyspepsia, flushing | Improves cardiopulmonary hemodynamics and exercise capacity36,37 | $17 for 20-mg tablet | $51 |
| Tadalafil (Adcirca) | 2.5 to 40 mg orally once per day (40 mg was needed for statistically significant effects38) | Headache, flushing, myalgias38 | Slowed incidence of clinical worsening and improved six-minute walk test38 Approved in the United States in 2009 for patients in functional classes II and III | $19 for 20-mg tablet | $38 for 40 mg per day |
FDA = U.S. Food and Drug Administration; NA = not applicable; PAH = pulmonary arterial hypertension.
*— Average wholesale cost based on Red Book. New York, NY: Thomson Reuters; 2009.
†—Currently not approved by the FDA (update 12/15/10: sitaxsentan was withdrawn from the market December 2010).
Endothelin is a potent vasoconstrictor and stimulator of vascular smooth muscle cell proliferation and is believed to be important in the pathophysiology of PAH.39 In the United States, currently available ERAs include ambrisentan (Letairis) and bosentan (Tracleer). A third ERA, sitaxsentan, is undergoing investigation pending approval from the U.S. Food and Drug Administration (update 12/15/10: sitaxsentan was withdrawn from the market December 2010).
Phosphodiesterase type 5 inhibitors increase the intracellular concentration of cyclic guanosine mono-phosphate, increase nitric oxide (which induces relaxation of blood vessels), and create antiproliferative effects on vascular smooth muscle cells. Sildenafil (Revatio) and tadalafil (Adcirca) are two phosphodi-esterase type 5 inhibitors currently approved for PAH treatment.
Because multiple pathophysiological mechanisms may be involved in PAH, combination therapy may be considered. The addition of sildenafil to epoprostenol monotherapy has been shown to safely improve pulmonary arterial pressure.40 More research is needed in this area. More research is also needed to determine whether patients with different types of PAH should have different treatment strategies.
INTERVENTIONAL PROCEDURES
Severe PAH may benefit from an interatrial defect by allowing right-to-left shunting and increasing systemic output. Balloon atrial septostomy has been evaluated only in small studies and the benefit is uncertain. Mortality rates for this procedure range from 5 to 15 percent.7 At this time, balloon atrial septostomy is recommended only for patients in advanced functional classes III and IV (Table 541 ) to relieve symptoms before lung transplantation or as a sole treatment modality when no other options are available.6
Table 5. Functional Assessment Classification of Patients with Pulmonary Hypertension
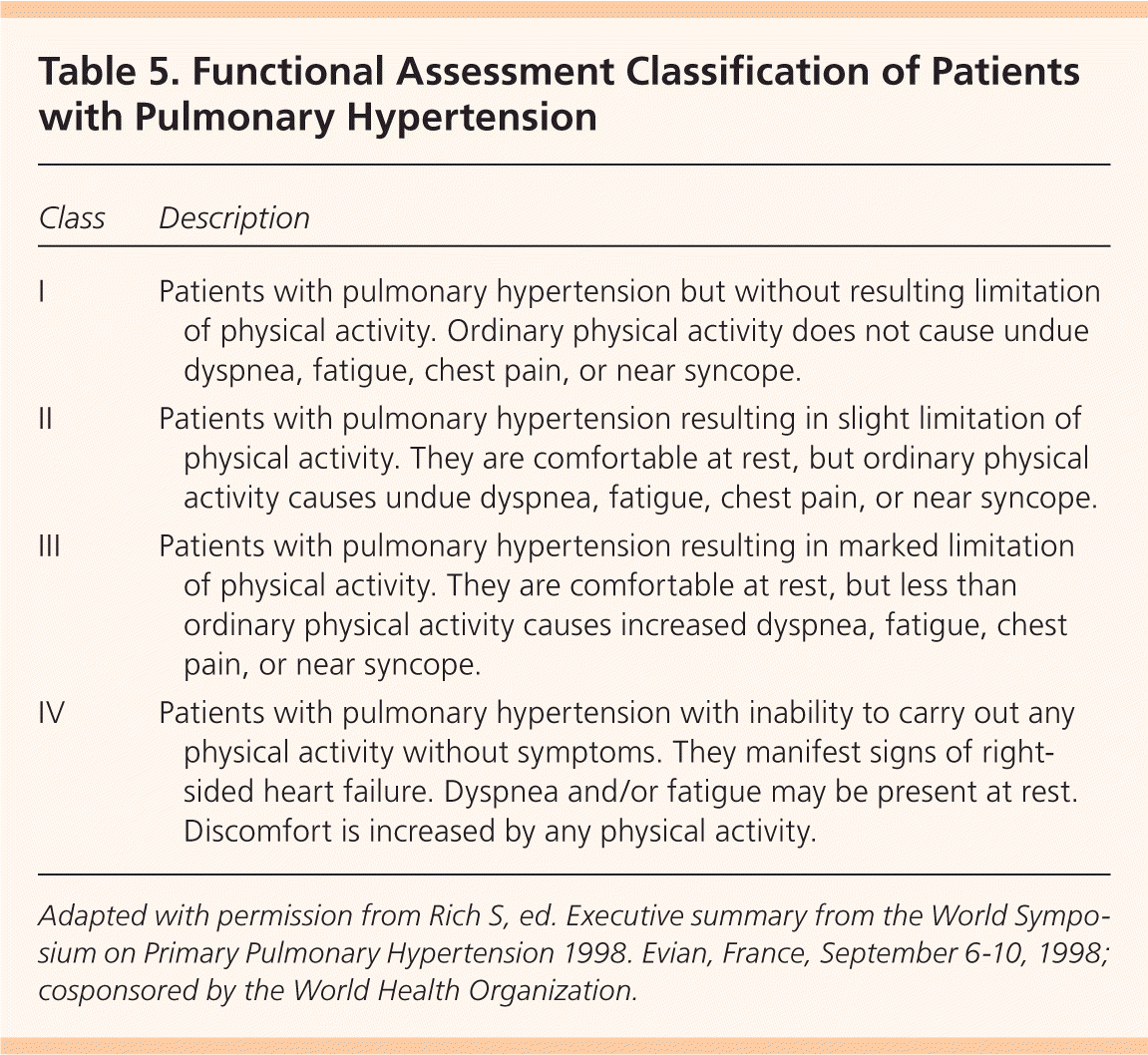
| Class | Description |
|---|---|
| I | Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnea, fatigue, chest pain, or near syncope. |
| II | Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest, but ordinary physical activity causes undue dyspnea, fatigue, chest pain, or near syncope. |
| III | Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest, but less than ordinary physical activity causes increased dyspnea, fatigue, chest pain, or near syncope. |
| IV | Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms. They manifest signs of right- sided heart failure. Dyspnea and/or fatigue may be present at rest. Discomfort is increased by any physical activity. |
Adapted with permission from Rich S, ed. Executive summary from the World Symposium on Primary Pulmonary Hypertension 1998. Evian, France, September 6–10, 1998; cosponsored by the World Health Organization.
Lung transplantation has been evaluated only in prospective uncontrolled series because formal randomized controlled trials are considered unethical in the absence of alternative treatment options. The three- and five-year survival rates after transplantation are 55 and 45 percent, respectively.6 Lung and heart-lung transplantation are indicated in patients in advanced functional classes III and IV who are refractory to available medical treatments.6 Compared with other lung transplant recipients, patients with PAH have a poorer postoperative course and a higher mortality rate.42 The unpredictability of the waiting list period and the donor organ shortage complicate the decision-making process.
Prognosis, Preventive Care, and Pregnancy
There is no cure for PAH, and the overall prognosis is poor. The course of PAH is progressive and irreversible, with an approximate mortality rate of 15 percent within one year, despite modern therapy.43 Lifestyle changes and appropriate therapy can relieve symptoms and potentially slow disease progression.
Patients with PAH should receive the pneumococcal vaccine and annual influenza immunization. Smoking cessation is recommended. A healthy lifestyle, including physical activity as tolerated and a nutritious diet, is also advised.
