The systemic vasculitides are characterized by inflammation of blood vessel walls. Vessels of any type, in any organ can be affected, resulting in a broad spectrum of signs and symptoms. The heterogenous nature of vasculitides presents a diagnostic challenge. The American College of Rheumatology classification criteria and the Chapel Hill Consensus Conference nomenclature are the most widely used to distinguish different forms of vasculitis. The Chapel Hill Consensus Conference nomenclature defines 10 primary vasculitides based on vessel size (large, medium, and small). The diagnosis relies on the recognition of a compatible clinical presentation supported by specific laboratory or imaging tests and confirmatory histology. Antineutrophilic cytoplasmic antibody testing has been of particular benefit in defining a subgroup of small vessel vasculitides. Treatment is based on clinical presentation and the pattern of organ involvement. Glucocorticoids are the primary treatment for many forms of vasculitis. Additional immunosuppressive agents, including methotrexate and cyclophosphamide, are sometimes required. Newer approaches, such as the use of anti-tumor necrosis factor or B cell therapies, are being tried in resistant cases. Patients can experience considerable treatment-related toxicity, especially infection from immunosuppressive therapy and adverse effects from steroids (e.g., osteoporosis, diabetes mellitus, cataract). Vitamin D and calcium prophylaxis are recommended in patients on long-term steroid therapy.
Vasculitis refers to a heterogenous group of disorders in which there is inflammation and damage in blood vessel walls, leading to tissue necrosis. These are relatively uncommon disorders, with a reported annual incidence of 40 to 54 cases per 1 million persons.1 The incidence appears to be affected by geography, age, and seasonal challenges. Vasculitis may be limited to skin or other organs, or may be a multisystem disorder with multiple manifestations.
SORT: KEY RECOMMENDATIONS FOR PRACTICE

| Clinical recommendation | Evidence rating | References |
|---|---|---|
| The American College of Rheumatology classification criteria and the Chapel Hill Consensus Conference nomenclature may be used to distinguish different forms of vasculitis. | C | 2–6 |
| The Birmingham Vasculitis Activity Score can be used to monitor the activity of the specific condition during treatment. | C | 9 |
| A definitive diagnosis of systemic vasculitis should be made by the presence of characteristic symptoms and signs of vasculitis and at least one of the following: histologic evidence of vasculitis, positive serology for antineutrophilic cytoplasmic antibody, or specific indirect evidence of vasculitis. | C | 10–16 |
| For patients with localized and early vasculitis, treatment with steroids and methotrexate or cyclophosphamide is recommended for induction of remission. | C | 18–20, 23 |
| Steroids and cyclophosphamide are recommended for initial treatment of generalized organ-threatening vasculitis. | C | 18–20 |
| Patients presenting with severe life-threatening vasculitis (severe renal failure or pulmonary hemorrhage) should be treated with cyclophosphamide (pulsed intravenous or continuous oral) and steroids, with adjuvant plasma exchange. | C | 18–23 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.xml.
Classification
Numerous classifications of vasculitis have been proposed. The American College of Rheumatology has classification criteria for seven primary vasculitides: polyarteritis nodosa, Churg-Strauss syndrome, Wegener granulomatosis, hypersensitivity vasculitis, Henoch-Schönlein purpura, giant cell arteritis, and Takayasu arteritis.2 Microscopic polyangiitis is not included in the classification criteria. The American College of Rheumatology criteria were designed for research studies, but are often used for diagnosis, and do not include antineutrophilic cytoplasmic antibody (ANCA) testing for the diagnosis of small vessel vasculitis or require biopsy or angiography for vasculitis classification. The criteria have poor reliability when applied as diagnostic criteria.3
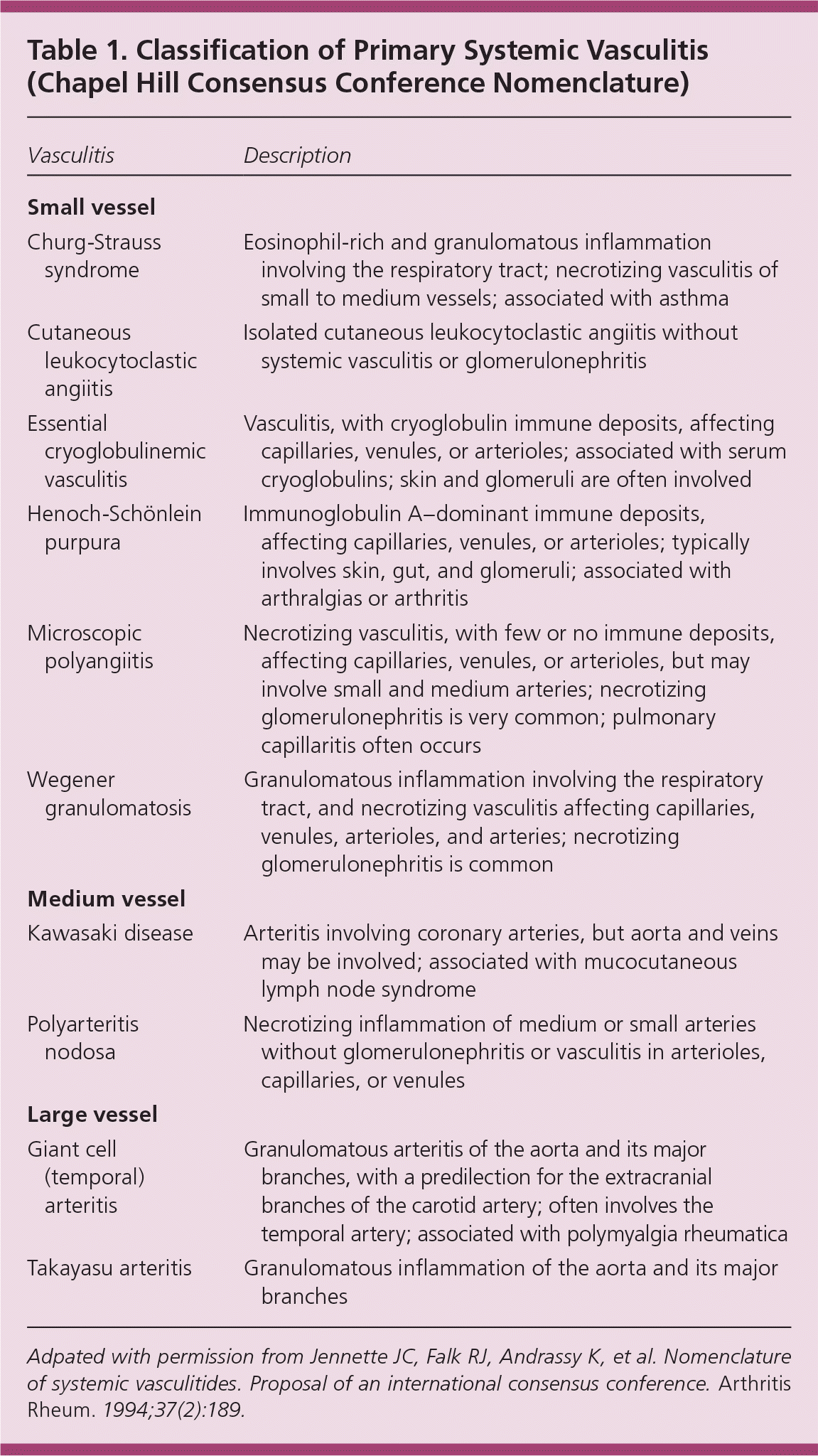
In 1994, the Chapel Hill Consensus Conference proposed a nomenclature defining 10 primary vasculitides based on vessel size (large, medium, and small; Table 1).4 The nomenclature includes microscopic polyangiitis and defines it as vasculitis involving microscopic vessels (arterioles, venules, and small capillaries). Microscopic polyangiitis is distinguished from polyarteritis nodosa by the presence of microscopic vessel involvement. Polyarteritis nodosa must have no involvement of microscopic vessels, including no glomerulonephritis. The nomenclature also stresses the importance of ANCA testing in the diagnosis of vasculitis, particularly in differentiating Wegener granulomatosis and microscopic polyangiitis in persons with pulmonary and renal involvement. The advantage of the Chapel Hill Consensus Conference nomenclature is its clarity and simplicity. However, the usefulness of this nomenclature as diagnostic criteria has been questioned.5,6
There is no recommended diagnostic standard for vasculitis. In the absence of validated diagnostic criteria, the American College of Rheumatology classification criteria and the Chapel Hill Consensus Conference nomenclature are most widely used in clinical practice to distinguish different forms of vasculitis.
Table 1. Classification of Primary Systemic Vasculitis (Chapel Hill Consensus Conference Nomenclature)
Pathogenesis
The pathogenesis of vasculitides is poorly understood. Three possible mechanisms of vascular damage are immune complex deposition, ANCAs (humoral response), and T-lymphocyte response with granuloma formation (cell-mediated).7,8 The end result of these various pathways is endothelial cell activation, with subsequent vessel obstruction and ischemia of dependent tissue. This may cause hemorrhage in the surrounding tissues and, in some cases, weakening of the vessel wall, which leads to the formation of aneurysms. For almost all forms of vasculitis, the triggering event initiating and driving this inflammatory response is unknown. Many small vessel vasculitides have a paucity of vascular immune deposits and, therefore, other mechanisms have been sought for these so-called pauci-immune vasculitides.
Clinical Features
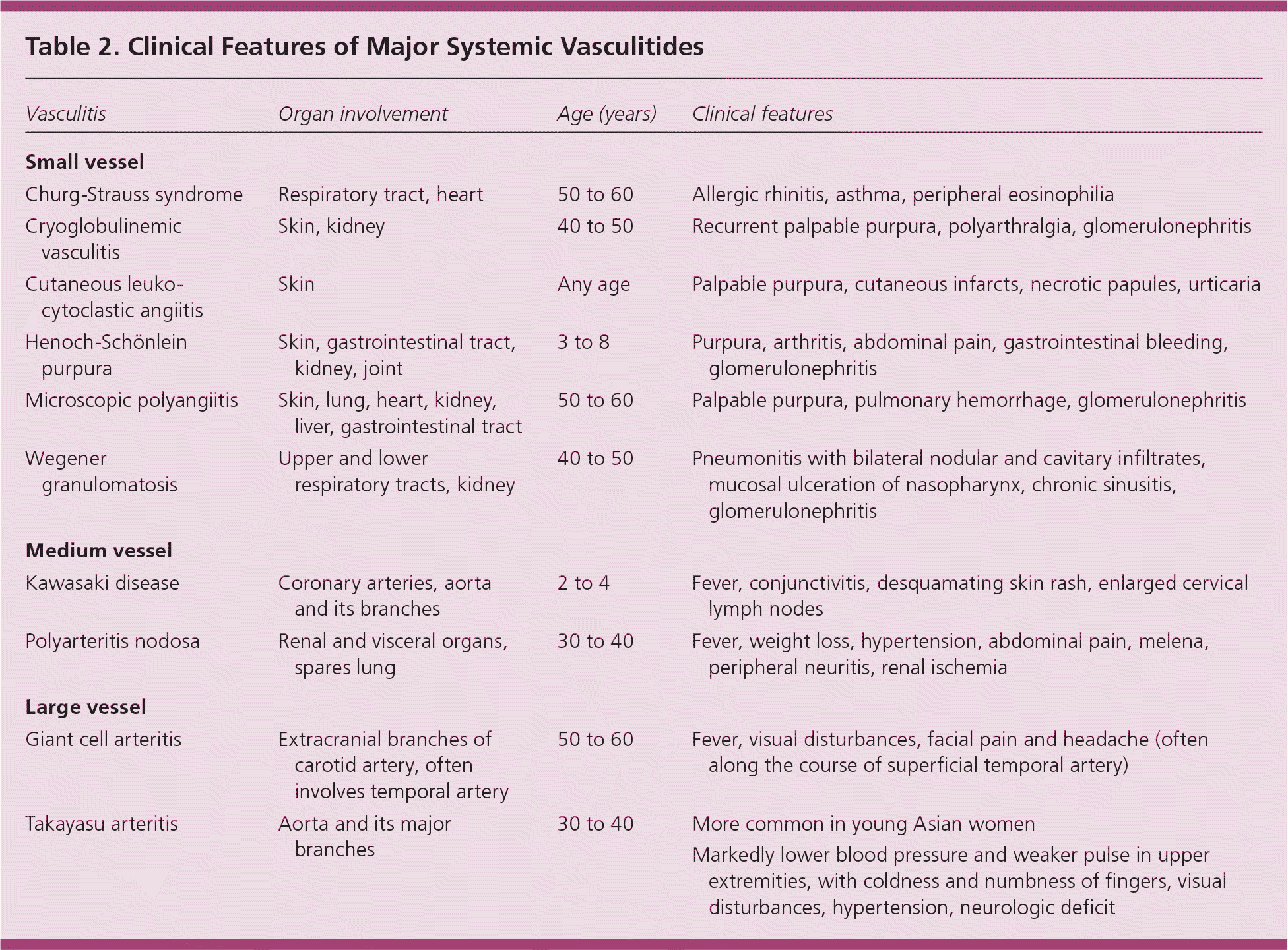
Patients with vasculitis typically have prodromal symptoms, constitutional disturbances, and organ-specific manifestations. Patients can present to the family physician with nonspecific signs or symptoms (e.g., fever, rash, myalgia, arthralgia, malaise, weight loss) or to the emergency department with life-threatening features (e.g., massive hemoptysis, renal failure). Manifestations vary, depending on the size, site, and extent of vessels involved. Clinical manifestations of various forms of vasculitis are discussed in Table 2.
Table 2. Clinical Features of Major Systemic Vasculitides
| Vasculitis | Organ involvement | Age (years) | Clinical features |
|---|---|---|---|
| Small vessel | |||
| Churg-Strauss syndrome | Respiratory tract, heart | 50 to 60 | Allergic rhinitis, asthma, peripheral eosinophilia |
| Cryoglobulinemic vasculitis | Skin, kidney | 40 to 50 | Recurrent palpable purpura, polyarthralgia, glomerulonephritis |
| Cutaneous leukocytoclastic angiitis | Skin | Any age | Palpable purpura, cutaneous infarcts, necrotic papules, urticaria |
| Henoch-Schönlein purpura | Skin, gastrointestinal tract, kidney, joint | 3 to 8 | Purpura, arthritis, abdominal pain, gastrointestinal bleeding, glomerulonephritis |
| Microscopic polyangiitis | Skin, lung, heart, kidney, liver, gastrointestinal tract | 50 to 60 | Palpable purpura, pulmonary hemorrhage, glomerulonephritis |
| Wegener granulomatosis | Upper and lower respiratory tracts, kidney | 40 to 50 | Pneumonitis with bilateral nodular and cavitary infiltrates, mucosal ulceration of nasopharynx, chronic sinusitis, glomerulonephritis |
| Medium vessel | |||
| Kawasaki disease | Coronary arteries, aorta and its branches | 2 to 4 | Fever, conjunctivitis, desquamating skin rash, enlarged cervical lymph nodes |
| Polyarteritis nodosa | Renal and visceral organs, spares lung | 30 to 40 | Fever, weight loss, hypertension, abdominal pain, melena, peripheral neuritis, renal ischemia |
| Large vessel | |||
| Giant cell arteritis | Extracranial branches of carotid artery, often involves temporal artery | 50 to 60 | Fever, visual disturbances, facial pain and headache (often along the course of superficial temporal artery) |
| Takayasu arteritis | Aorta and its major branches | 30 to 40 | More common in young Asian women |
| Markedly lower blood pressure and weaker pulse in upper extremities, with coldness and numbness of fingers, visual disturbances, hypertension, neurologic deficit | |||
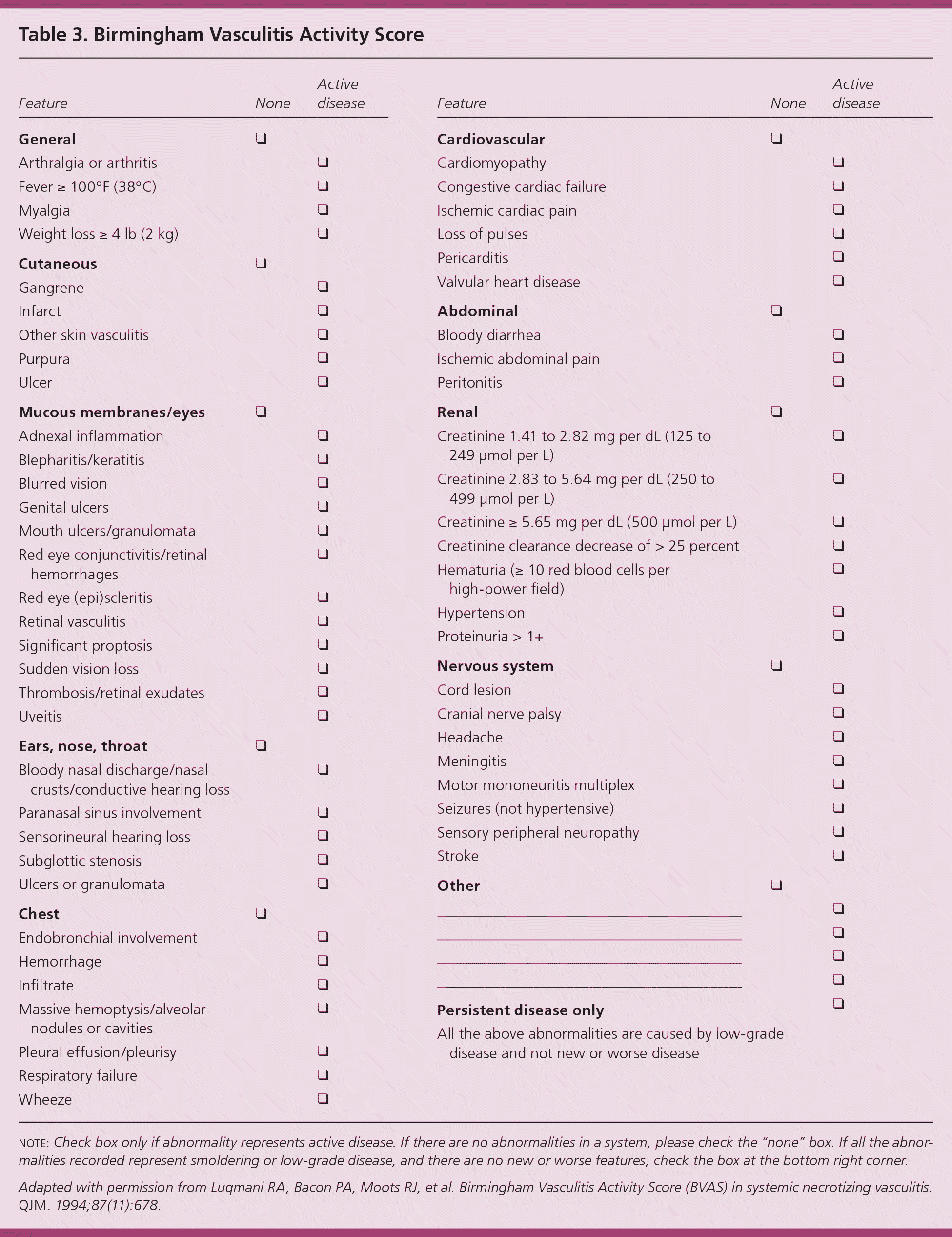
The Birmingham Vasculitis Activity Score is a checklist of signs and symptoms that are consistent with the diagnosis of systemic vasculitis and can be used to monitor the activity of the specific condition during treatment (Table 3).9
Table 3. Birmingham Vasculitis Activity Score
| Feature | None | Active disease |
|---|---|---|
| General | □ | |
| Arthralgia or arthritis | □ | |
| Fever ≥ 100°F (38°C) | □ | |
| Myalgia | □ | |
| Weight loss ≥ 4 lb (2 kg) | □ | |
| Cutaneous | □ | |
| Gangrene | □ | |
| Infarct | □ | |
| Other skin vasculitis | □ | |
| Purpura | □ | |
| Ulcer | □ | |
| Mucous membranes/eyes | □ | |
| Adnexal inflammation | □ | |
| Blepharitis/keratitis | □ | |
| Blurred vision | □ | |
| Genital ulcers | □ | |
| Mouth ulcers/granulomata | □ | |
| Red eye conjunctivitis/retinal hemorrhages | □ | |
| Red eye (epi)scleritis | □ | |
| Retinal vasculitis | □ | |
| Significant proptosis | □ | |
| Sudden vision loss | □ | |
| Thrombosis/retinal exudates | □ | |
| Uveitis | □ | |
| Ears, nose, throat | □ | |
| Bloody nasal discharge/nasal crusts/conductive hearing loss | □ | |
| Paranasal sinus involvement | □ | |
| Sensorineural hearing loss | □ | |
| Subglottic stenosis | □ | |
| Ulcers or granulomata | □ | |
| Chest | □ | |
| Endobronchial involvement | □ | |
| Hemorrhage | □ | |
| Infiltrate | □ | |
| Massive hemoptysis/alveolar nodules or cavities | □ | |
| Pleural effusion/pleurisy | □ | |
| Respiratory failure | □ | |
| Wheeze | □ | |
| Cardiovascular | □ | |
| Cardiomyopathy | □ | |
| Congestive cardiac failure | □ | |
| Ischemic cardiac pain | □ | |
| Loss of pulses | □ | |
| Pericarditis | □ | |
| Valvular heart disease | □ | |
| Abdominal | □ | |
| Bloody diarrhea | □ | |
| Ischemic abdominal pain | □ | |
| Peritonitis | □ | |
| Renal | □ | |
| Creatinine 1.41 to 2.82 mg per dL (125 to 249 μmol per L) | □ | |
| Creatinine 2.83 to 5.64 mg per dL (250 to 499 μmol per L) | □ | |
| Creatinine ≥ 5.65 mg per dL (500 μmol per L) | □ | |
| Creatinine clearance decrease of > 25 percent | □ | |
| Hematuria (≥ 10 red blood cells per high-power field) | □ | |
| Hypertension | □ | |
| Proteinuria > 1+ | □ | |
| Nervous system | □ | |
| Cord lesion | □ | |
| Cranial nerve palsy | □ | |
| Headache | □ | |
| Meningitis | □ | |
| Motor mononeuritis multiplex | □ | |
| Seizures (not hypertensive) | □ | |
| Sensory peripheral neuropathy | □ | |
| Stroke | □ | |
| Other | □ | |
| ___________________ | □ | |
| ___________________ | □ | |
| ___________________ | □ | |
| ___________________ | □ | |
| Persistent disease only All the above abnormalities are caused by low-grade disease and not new or worse disease | □ |
note: Check box only if abnormality represents active disease. If there are no abnormalities in a system, please check the “none” box. If all the abnormalities recorded represent smoldering or low-grade disease, and there are no new or worse features, check the box at the bottom right corner.
Adapted with permission from Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87(11):678.
General Diagnostic Approach
The primary systemic vasculitides are difficult to diagnose because the clinical manifestations mimic several infectious, neoplastic, and autoimmune conditions (Table 4). Attempts should first be made to exclude the malignant and infectious processes. The patient's age, sex, and demographic or ethnic origin are also important. Finally, type and extent of organ involvement and the size of the vessels involved should be determined. Certain organ-specific symptoms, not otherwise explained, may be clues leading to a more specific diagnosis (Table 2). A definitive diagnosis of systemic vasculitis should be made by the presence of characteristic symptoms and signs of vasculitis and at least one of the following: histologic evidence of vasculitis; positive serology for ANCA; or specific indirect evidence of vasculitis.10–16 It is important to ascertain that no other diagnosis accounts for the presenting symptoms and signs.
Table 4. Selected Conditions Mimicking Primary Systemic Vasculitis

| Antiphospholipid syndrome |
| Atheroembolic disease |
| Atheromatous vascular disease |
| Cocaine and amphetamine abuse |
| Hypersensitivity reactions |
| Infective endocarditis |
| Multiple myeloma |
| Paraneoplastic syndromes |
| Secondary causes of vasculitis (rheumatoid arthritis, systemic lupus erythematosus, scleroderma, hepatitis B and C infection, lymphoma, and solid organ malignancy) |
| Sickle cell disease |
Laboratory Testing
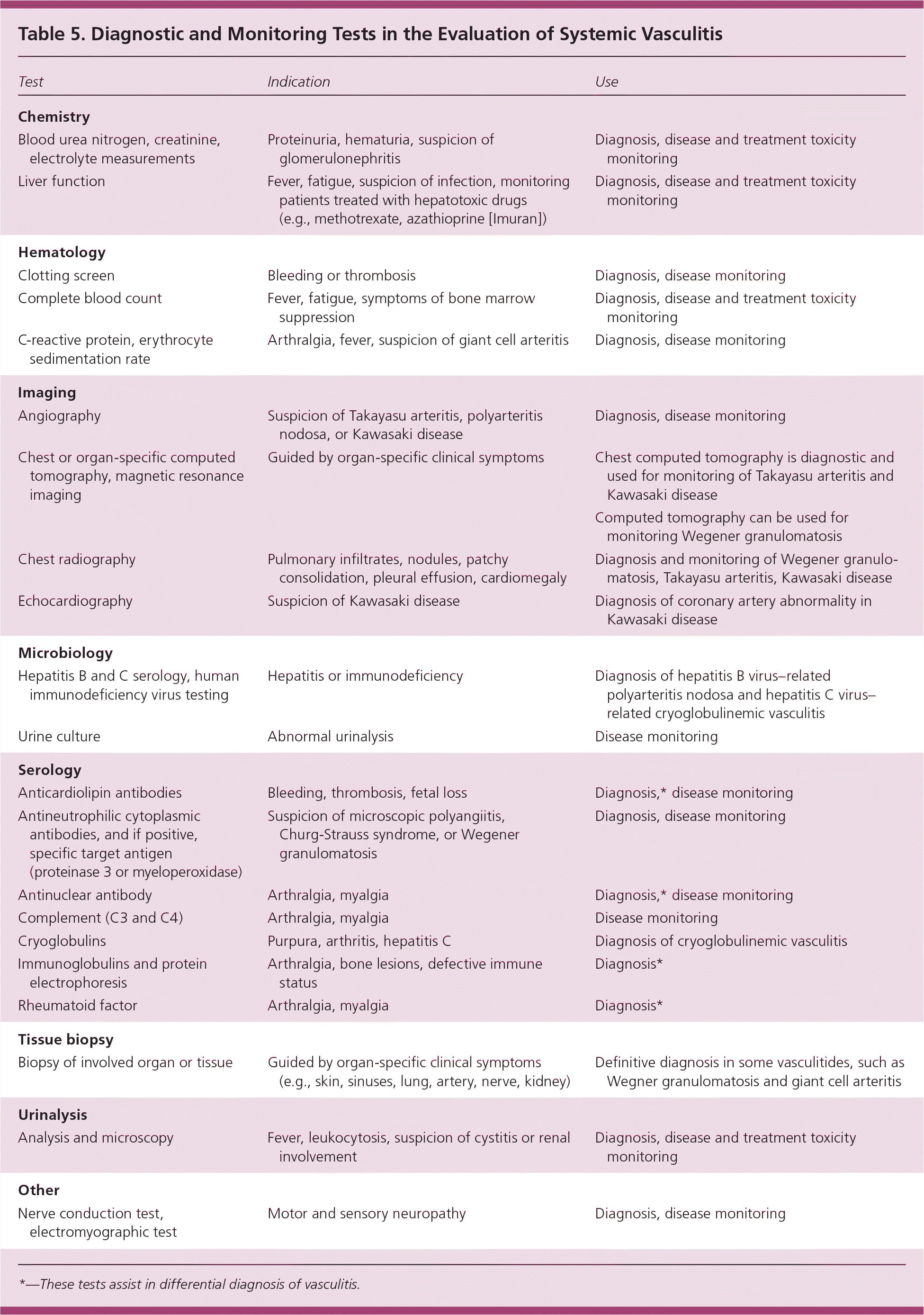
Laboratory testing is important to determine the organs involved, to exclude other diseases, and to monitor the disease and treatment toxicity. A typical plan of investigation for a patient with vasculitis is summarized in Table 5.
Table 5. Diagnostic and Monitoring Tests in the Evaluation of Systemic Vasculitis
| Test | Indication | Use |
|---|---|---|
| Chemistry | ||
| Blood urea nitrogen, creatinine, electrolyte measurements | Proteinuria, hematuria, suspicion of glomerulonephritis | Diagnosis, disease and treatment toxicity monitoring |
| Liver function | Fever, fatigue, suspicion of infection, monitoring patients treated with hepatotoxic drugs (e.g., methotrexate, azathioprine [Imuran]) | Diagnosis, disease and treatment toxicity monitoring |
| Hematology | ||
| Clotting screen | Bleeding or thrombosis | Diagnosis, disease monitoring |
| Complete blood count | Fever, fatigue, symptoms of bone marrow suppression | Diagnosis, disease and treatment toxicity monitoring |
| C-reactive protein, erythrocyte sedimentation rate | Arthralgia, fever, suspicion of giant cell arteritis | Diagnosis, disease monitoring |
| Imaging | ||
| Angiography | Suspicion of Takayasu arteritis, polyarteritis nodosa, or Kawasaki disease | Diagnosis, disease monitoring |
| Chest or organ-specific computed tomography, magnetic resonance imaging | Guided by organ-specific clinical symptoms | Chest computed tomography is diagnostic and used for monitoring of Takayasu arteritis and Kawasaki disease |
| Computed tomography can be used for monitoring Wegener granulomatosis | ||
| Chest radiography | Pulmonary infiltrates, nodules, patchy consolidation, pleural effusion, cardiomegaly | Diagnosis and monitoring of Wegener granulomatosis, Takayasu arteritis, Kawasaki disease |
| Echocardiography | Suspicion of Kawasaki disease | Diagnosis of coronary artery abnormality in Kawasaki disease |
| Microbiology | ||
| Hepatitis B and C serology, human immunodeficiency virus testing | Hepatitis or immunodeficiency | Diagnosis of hepatitis B virus–related polyarteritis nodosa and hepatitis C virus–related cryoglobulinemic vasculitis |
| Urine culture | Abnormal urinalysis | Disease monitoring |
| Serology | ||
| Anticardiolipin antibodies | Bleeding, thrombosis, fetal loss | Diagnosis,* disease monitoring |
| Antineutrophilic cytoplasmic antibodies, and if positive, specific target antigen(proteinase 3 or myeloperoxidase) | Suspicion of microscopic polyangiitis, Churg-Strauss syndrome, or Wegener granulomatosis | Diagnosis, disease monitoring |
| Antinuclear antibody | Arthralgia, myalgia | Diagnosis,* disease monitoring |
| Complement (C3 and C4) | Arthralgia, myalgia | Disease monitoring |
| Cryoglobulins | Purpura, arthritis, hepatitis C | Diagnosis of cryoglobulinemic vasculitis |
| Immunoglobulins and protein electrophoresis | Arthralgia, bone lesions, defective immune status | Diagnosis* |
| Rheumatoid factor | Arthralgia, myalgia | Diagnosis* |
| Tissue biopsy | ||
| Biopsy of involved organ or tissue | Guided by organ-specific clinical symptoms(e.g., skin, sinuses, lung, artery, nerve, kidney) | Definitive diagnosis in some vasculitides, such as Wegner granulomatosis and giant cell arteritis |
| Urinalysis | ||
| Analysis and microscopy | Fever, leukocytosis, suspicion of cystitis or renal involvement | Diagnosis, disease and treatment toxicity monitoring |
| Other | ||
| Nerve conduction test, electromyographic test | Motor and sensory neuropathy | Diagnosis, disease monitoring |
*—These tests assist in differential diagnosis of vasculitis.
COMPLETE BLOOD COUNT
Patients with active vasculitis often have leukocytosis, anemia, and thrombocytopenia. Eosinophilia is a prominent feature in Churg-Strauss syndrome. A complete blood count is important to look for the bone marrow suppression that may result from vasculitis treatment.
ACUTE PHASE REACTANTS
An increased erythrocyte sedimentation rate and an elevated C-reactive protein level are common in patients with vasculitis, but are nonspecific and can occur in many settings, particularly infection. A normal erythrocyte sedimentation rate and C-reactive protein level can occur in vasculitis when the disease is inactive, and should not exclude the diagnosis. In patients with giant cell arteritis, an increased erythrocyte sedimentation rate can suggest the diagnosis and can be useful for disease monitoring when combined with compatible clinical features.
RENAL FUNCTION TESTS AND URINALYSIS
Blood urea nitrogen and serum creatinine measurements should be obtained, and urinalysis should be performed in any patient with suspected vasculitis. Proteinuria and hematuria suggest the possibility of glomerulonephritis. Monitoring creatinine and the urinalysis is useful to detect changes in disease activity. It may be helpful to identify bladder toxicity in patients treated with cyclophosphamide.
LIVER FUNCTION TESTS
Serum bilirubin and liver enzyme levels (aspartate and alanine transaminase, alkaline phosphatase, γ-glutamyltransferase) can provide clues for vasculitis that affects the liver, such as polyarteritis nodosa. Serial liver function tests are also important in monitoring patients treated with hepatotoxic drugs, such as methotrexate and azathioprine (Imuran).
ANTINEUTROPHIL CYTOPLASMIC ANTIBODIES
Serum from many patients with vasculitis shows the presence of ANCAs, which are a heterogeneous group of autoantibodies directed against enzymes found in neutrophils. There are two types of ANCAs based on indirect immunofluorescence patterns: cytoplasmic and perinuclear. The most common corresponding antigens identified by enzyme immunoassay are proteinase 3 for cytoplasmic ANCA and myeloperoxidase for perinuclear ANCA. All positive ANCA results based on immunofluorescence should be confirmed by enzyme immunoassay.10,11
The disorders characterized by circulating ANCAs are called ANCA-associated vasculitides, and include Churg-Strauss syndrome, microscopic polyangiitis, and Wegener granulomatosis. Either type of ANCA may occur in a patient with ANCA-associated small vessel vasculitis, but cytoplasmic ANCA is typically found in Wegener granulomatosis, and perinuclear ANCA is typically found in microscopic polyangiitis and Churg-Strauss syndrome. ANCAs are useful quantitative markers for these conditions, and the level reflects the degree of inflammation.12 As such, ANCAs increase during recurrence and are useful in monitoring response to treatment.13,14 Persistence of ANCAs is a predictor of poor response to treatment. Approximately 10 percent of patients with Wegener granulomatosis or microscopic polyangiitis have negative assays for ANCAs; therefore, a negative result does not completely rule out these diseases.17 In addition, ANCAs have been reported in other conditions, such as infections, inflammatory bowel disease, and drug-induced vasculitis.
Imaging
CHEST RADIOGRAPHY
Nonspecific abnormalities that can be seen on chest radiography include infiltrates, nodules, patchy consolidation, pleural effusion, and cardiomegaly. These findings can occur in many settings, but if unexplained, may raise the suspicion of vasculitis.
ANGIOGRAPHY
Angiography can show vascular occlusion and aneurysm. A diagnosis of polyarteritis nodosa can be confirmed by detection of aneurysms in mesenteric and renal arteries. Although conventional angiography is still accepted as the recommended diagnostic modality, computed tomography angiography and magnetic resonance angiography may be superior because they can provide valuable information regarding intraluminal pathology and thickening of the vessel wall. These techniques have been used for diagnosis and monitoring of Takayasu arteritis and Kawasaki disease.
ECHOCARDIOGRAPHY
Transthoracic echocardiography detects coronary artery abnormality in Kawasaki disease. Approximately 40 percent of children with Kawasaki disease have coronary artery lesions (ectasia or aneurysm) on echocardiography.15 Echocardiography is used for assessing coronary artery blood flow and the degree of coronary stenosis in patients with Takayasu arteritis.16
ULTRASONOGRAPHY
Ultrasonography may be useful for diagnosis and monitoring of large vessel vasculitis. Patients with giant cell arteritis may have stenosis, occlusion, or halo sign (a dark area around the artery from vessel wall edema) of the superficial temporal artery.
COMPUTED TOMOGRAPHY
Computed tomography is of diagnostic value in patients with sinonasal Wegener granulomatosis. Findings include thickening of nasal mucosa and punctate bony destruction, mainly in the midline. Nodules or masses can be seen on chest computed tomography in approximately 90 percent of patients with Wegener granulomatosis.18
Other Testing
NERVE CONDUCTION TESTING
Motor and sensory neuropathy can occur in the context of systemic vasculitis when the vasa nervorum are affected (e.g., polyarteritis nodosa, Wegener granulomatosis, Churg-Strauss syndrome). Nerve conduction testing should be used for the evaluation of neurologic manifestations.
TISSUE BIOPSY
The definitive diagnosis of vasculitis is established by biopsy of the involved tissue (e.g., skin, sinuses, lung, artery, nerve, kidney), which determines the pattern of vessel inflammation. The presence of immunoglobulins and complement found by immunofluorescence on the tissue section may be helpful in elucidating the specific type of vasculitis. Biopsies are particularly valuable in ruling out other causes, but a negative biopsy does not rule out vasculitis.
Treatment
The initial presentation may be to the family physician or any number of subspecialists, including a dermatologist, rheumatologist, nephrologist, otolaryngologist, or pulmonologist. Most patients are treated by a rheumatologist experienced with these diseases. The rheumatologist works with the patient's family physician, who monitors the disease progression and complications of treatment.
The treatment includes three phases: induction of remission, maintenance, and treatment of relapse. The severity and extent of the disease divides patients into three groups: those with localized or early disease, those with generalized disease with threatened organ involvement, and those with severe or life-threatening disease. For patients with localized and early disease, treatment with steroids and methotrexate or cyclophosphamide is recommended for induction of remission.18–20 Methotrexate may be associated with a higher relapse rate.21,22 Evidence of relapse or disease progression despite treatment with methotrexate requires the use of cyclophosphamide. Initial treatment of generalized organ-threatening disease should include steroids and cyclophosphamide.18–20 Cyclophosphamide can be administered as an intravenous infusion every two weeks (and later every three weeks), or as a daily low-dose oral treatment. There is no difference in remission rates or relapse risk between oral and intravenous regimens. Steroids are given as daily oral prednisone (1 mg per kg, up to 60 mg daily). Pulsed intravenous steroids can be given just before or with the first two intravenous pulses of cyclophosphamide. Patients presenting with severe life-threatening disease (severe renal failure or pulmonary hemorrhage) should be treated with cyclophosphamide (pulsed intravenous or continuous oral) and steroids, with adjuvant plasma exchange.19,23
Maintenance therapy with either azathioprine or methotrexate is initiated if remission has occurred after three to six months of induction therapy. Steroid dosage is tapered during this phase. Patients may need to continue maintenance treatment for up to 24 months.24 Maintenance treatment for up to five years is recommended in patients with Wegener granulomatosis and patients who remain ANCA-positive.19 Some patients may require treatment indefinitely. Disease relapse may occur anytime after the remission. Serial measurements of ANCA are not closely associated with disease activity; therefore, treatment should not be solely guided on the basis of an increase in ANCA.25 Relapsing disease can be managed with an increase in steroid dose, optimization of the current immunosuppressant, or combination of an immunosuppressant with an increased dose of steroid.
Novel biologic therapies targeted against specific components of the immune system are being used for systemic vasculitis, particularly for patients in whom conventional therapy has failed. Agents such as infliximab (Remicade; human chimeric anti-tumor necrosis factor [TNF]-α monoclonal antibody), etanercept (Enbrel; fusion protein of the p75 TNF-α receptor and immunoglobulin G1), adalimumab (Humira; fully humanized IgG1 anti-TNF-α monoclonal antibody), rituximab (Rituxan; anti-CD20 chimeric mouse/human monoclonal antibody), anakinra (Kineret; recombinant interleukin-1 receptor antagonist), and intravenous immune globulins may be used in refractory disease.26,27
Patients with systemic vasculitis are at increased risk of comorbidities resulting from disease-related end organ damage and immunosuppressive therapy. The immunosuppressive medications used for the treatment of systemic vasculitis cause serious adverse effects during the first year of therapy. Steroids and cyclophosphamide predispose patients to life-threatening infections. Cyclophosphamide can cause hemorrhagic cystitis, ovarian and testicular failure, and bladder cancer. Diagnosis and treatment of these complications are coordinated with the family physician. Recommendations regarding detecting and preventing these complications include use of mesna (Mesnex) for protecting against urothelial toxicity of cyclophosphamide, antifungal prophylaxis, prophylaxis against Pneumocystis jiroveci, consideration for Staphylococcus aureus treatment, screening for cervical malignancy, and counseling about infertility with cyclophosphamide. Adverse effects of long-term steroid use (e.g., diabetes mellitus, osteoporosis, cataract) should be assessed. Vitamin D and calcium prophylaxis are recommended in patients on long-term therapy with steroids. Table 6 summarizes the drugs and treatments for systemic vasculitis.23
Table 6. Summary of Drugs and Treatments Used for Systemic Vasculitis
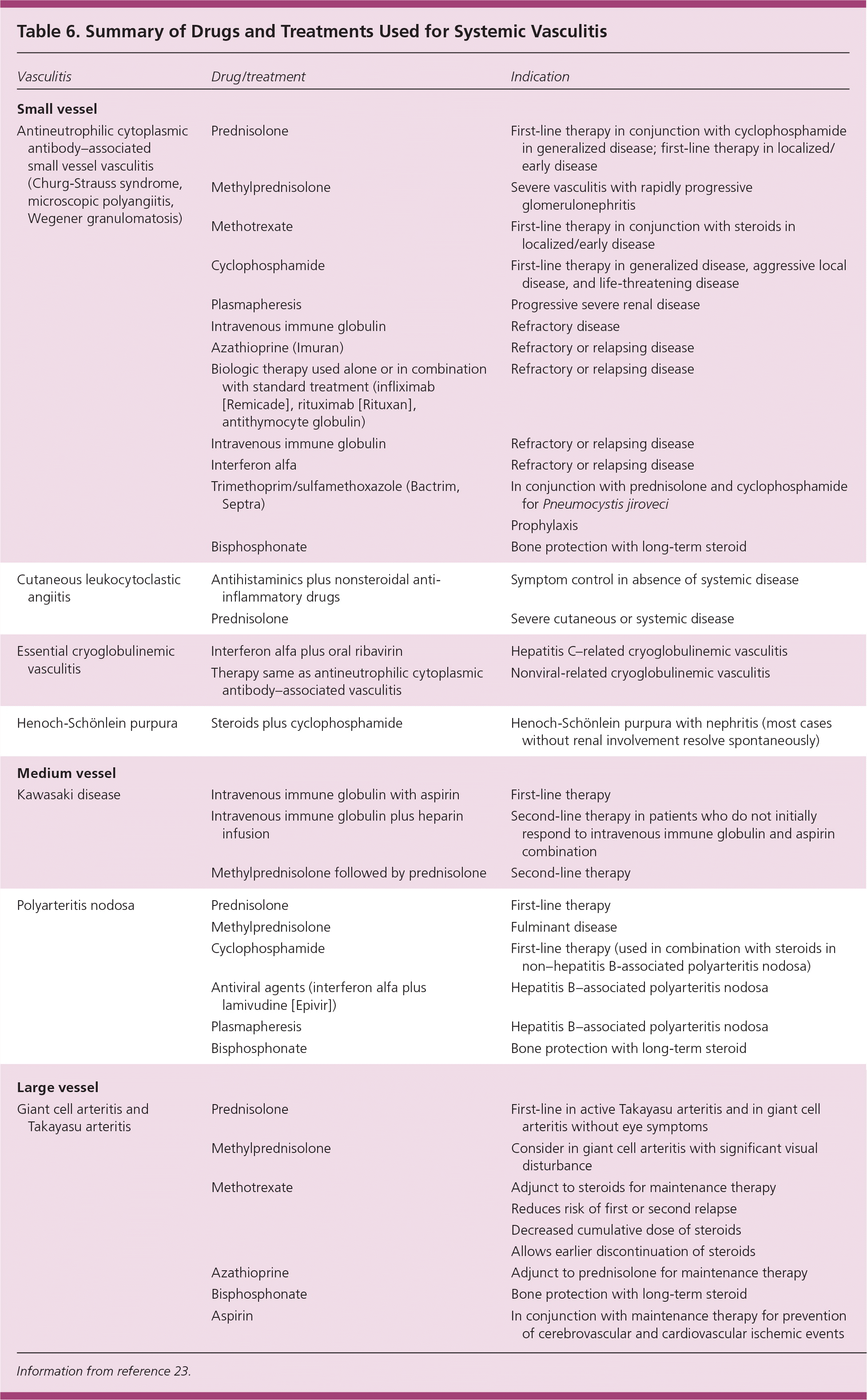
| Vasculitis | Drug/treatment | Indication |
|---|---|---|
| Small vessel | ||
| Antineutrophilic cytoplasmic antibody–associated small vessel vasculitis (Churg-Strauss syndrome, microscopic polyangiitis, Wegener granulomatosis) | Prednisolone | First-line therapy in conjunction with cyclophosphamide in generalized disease; first-line therapy in localized/early disease |
| Methylprednisolone | Severe vasculitis with rapidly progressive glomerulonephritis | |
| Methotrexate | First-line therapy in conjunction with steroids in localized/early disease | |
| Cyclophosphamide | First-line therapy in generalized disease, aggressive local disease, and life-threatening disease | |
| Plasmapheresis | Progressive severe renal disease | |
| Intravenous immune globulin | Refractory disease | |
| Azathioprine (Imuran) | Refractory or relapsing disease | |
| Biologic therapy used alone or in combination with standard treatment (infliximab [Remicade], rituximab [Rituxan], antithymocyte globulin) | Refractory or relapsing disease | |
| Intravenous immune globulin | Refractory or relapsing disease | |
| Interferon alfa | Refractory or relapsing disease | |
| Trimethoprim/sulfamethoxazole (Bactrim, Septra) | In conjunction with prednisolone and cyclophosphamide for Pneumocystis jiroveci | |
| Prophylaxis | ||
| Bisphosphonate | Bone protection with long-term steroid | |
| Cutaneous leukocytoclastic angiitis | Antihistaminics plus nonsteroidal anti-inflammatory drugs | Symptom control in absence of systemic disease |
| Prednisolone | Severe cutaneous or systemic disease | |
| Essential cryoglobulinemic vasculitis | Interferon alfa plus oral ribavirin | Hepatitis C–related cryoglobulinemic vasculitis |
| Therapy same as antineutrophilic cytoplasmic antibody–associated vasculitis | Nonviral-related cryoglobulinemic vasculitis | |
| Henoch-Schönlein purpura | Steroids plus cyclophosphamide | Henoch-Schönlein purpura with nephritis (most cases without renal involvement resolve spontaneously) |
| Medium vessel | ||
| Kawasaki disease | Intravenous immune globulin with aspirin | First-line therapy |
| Intravenous immune globulin plus heparin infusion | Second-line therapy in patients who do not initially respond to intravenous immune globulin and aspirin combination | |
| Methylprednisolone followed by prednisolone | Second-line therapy | |
| Polyarteritis nodosa | Prednisolone | First-line therapy |
| Methylprednisolone | Fulminant disease | |
| Cyclophosphamide | First-line therapy (used in combination with steroids in non–hepatitis B-associated polyarteritis nodosa) | |
| Antiviral agents (interferon alfa plus lamivudine [Epivir]) | Hepatitis B–associated polyarteritis nodosa | |
| Plasmapheresis | Hepatitis B–associated polyarteritis nodosa | |
| Bisphosphonate | Bone protection with long-term steroid | |
| Large vessel | ||
| Giant cell arteritis and Takayasu arteritis | Prednisolone | First-line in active Takayasu arteritis and in giant cell arteritis without eye symptoms |
| Methylprednisolone | Consider in giant cell arteritis with significant visual disturbance | |
| Methotrexate | Adjunct to steroids for maintenance therapy | |
| Reduces risk of first or second relapse | ||
| Decreased cumulative dose of steroids | ||
| Allows earlier discontinuation of steroids | ||
| Azathioprine | Adjunct to prednisolone for maintenance therapy | |
| Bisphosphonate | Bone protection with long-term steroid | |
| Aspirin | In conjunction with maintenance therapy for prevention of cerebrovascular and cardiovascular ischemic events | |
Information from reference 23.
Management of systemic vasculitis is complicated. Educating patients about signs and symptoms, and monitoring typical adverse effects are helpful. Many patients will have a relatively benign, self-limited course, especially if the disease is limited to the skin; however, for patients with aggressive disease, such as ANCA-associated small vessel vasculitis, it is imperative to begin treatment without delay. The multisystem involvement in systemic vasculitis necessitates a multidisciplinary team approach to patient care. Recent advances in therapy have led to considerably better outcomes in patients with vasculitis.
