Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited cause of kidney disease. Enlarging cysts within the kidneys are the clinical hallmark of the disease. Renal manifestations include varying degrees of kidney injury, urinary tract infections, kidney stones, and hematuria. Extrarenal manifestations can include pain, hypertension, left ventricular hypertrophy, hepatic cysts, intracranial aneurysm, diverticulosis, and abdominal and inguinal hernias. The progression of ADPKD cannot be reversed with current treatment modalities; therefore, therapies target the resulting clinical manifestations. Early detection and management of hypertension are important to delay the progression of renal dysfunction and development of cardiovascular complications. Pain management includes evaluation of concomitant illnesses, use of analgesics, and adjuvant therapy. Fluoroquinolones may be the most useful class of antibiotics for the treatment of urinary tract infections because of their lipophilic properties and bactericidal action against gram-negative pathogens. Nephrolithiasis is twice as common in persons with ADPKD compared with the general population and is suggested by flank pain with or without hematuria. Cystic hemorrhages usually resolve within one week, although microscopic hematuria may still be present. Because of the proliferative effect of estrogen on hepatic cysts, oral contraceptives containing estrogen and menopausal estrogen therapy should be administered at the lowest effective dose or avoided in patients with ADPKD. Intracranial aneurysms are at least twice as common in patients with ADPKD than in the general population. Renal ultrasonography is the diagnostic modality of choice to screen at-risk individuals for ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited cause of kidney disease. ADPKD affects approximately 300,000 to 600,000 individuals nationwide without gender or racial predisposition,1 and it accounts for approximately 4.7% of cases of end-stage renal disease in the United States.2 It is a systemic disorder that causes cysts to develop in the kidneys (Figure 1) and in other areas of the body, leading to many clinical manifestations.
SORT: KEY RECOMMENDATIONS FOR PRACTICE

| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are recommended for initial hypertension management in patients with ADPKD. | C | 6, 11–13 |
| In patients with ADPKD who develop chronic kidney disease, the blood pressure goal is less than 140/90 mm Hg. | C | 13 |
| Fluoroquinolones, third-generation cephalosporins, and trimethoprim/sulfamethoxazole are effective for treating urinary tract infections and infected renal cysts in patients with ADPKD. Fluoroquinolones may be preferred given their lipophilic properties and bactericidal action against gram-negative pathogens. | C | 15, 17–20 |
| If overt urinary bleeding in a patient with ADPKD lasts longer than one week or occurs for the first time in a patient older than 50 years, he or she should be evaluated for neoplasm. | C | 6 |
| Because estrogen has a proliferative effect on hepatic cysts, oral contraceptives containing estrogen and menopausal estrogen therapy should be administered at the lowest effective dose or avoided in patients with ADPKD. | C | 1, 6, 27 |
ADPKD = autosomal dominant polycystic kidney disease.
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
BEST PRACTICES IN NEPHROLOGY: RECOMMENDATIONS FROM THE CHOOSING WISELY CAMPAIGN
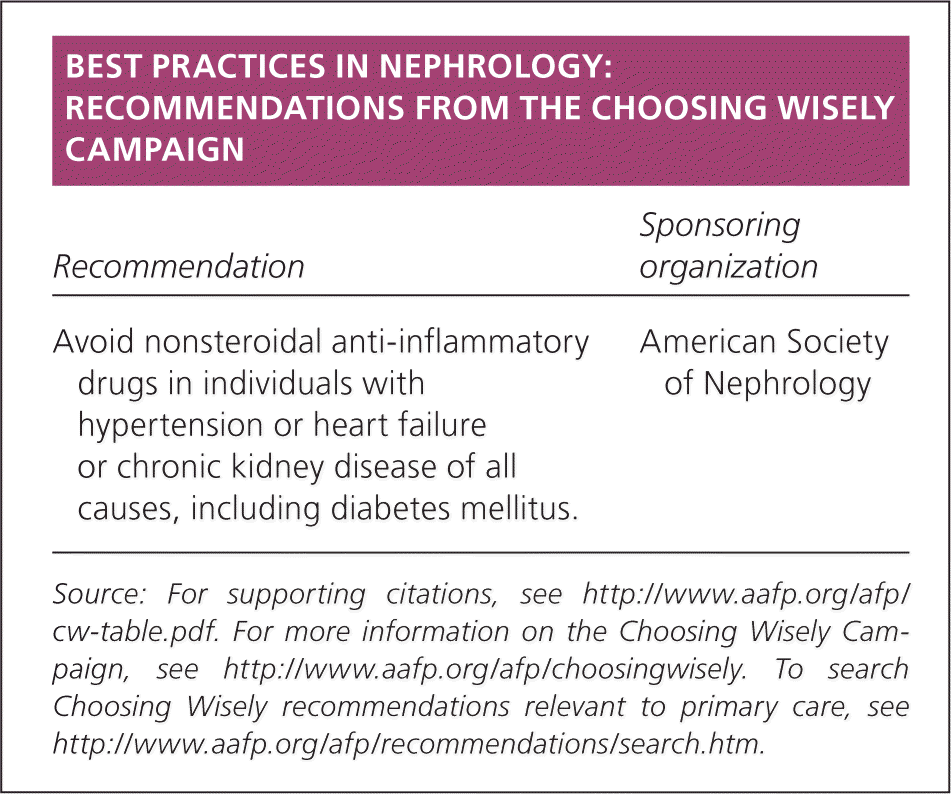
| Recommendation | Sponsoring organization |
|---|---|
| Avoid nonsteroidal anti-inflammatory drugs in individuals with hypertension or heart failure or chronic kidney disease of all causes, including diabetes mellitus. | American Society of Nephrology |
Source: For supporting citations, see https://www.aafp.org/afp/cw-table.pdf. For more information on the Choosing Wisely Campaign, see https://www.aafp.org/afp/choosingwisely. To search Choosing Wisely recommendations relevant to primary care, see https://www.aafp.org/afp/recommendations/search.htm.
Figure 1.
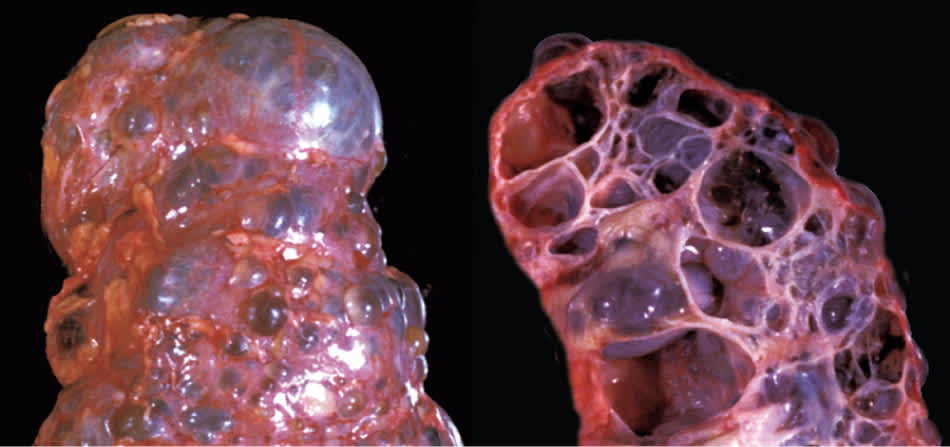
External surface and internal cut of a kidney from a patient with autosomal dominant polycystic kidney disease.
Copyright © J. Charles Jennette, MD, Department of Pathology and Laboratory Medicine, University of North Carolina.
Etiology and Pathogenesis
ADPKD is a heterogenetic disorder caused by mutations in the PKD1 gene located on chromosome 16 or in the PKD2 gene located on chromosome 4. The PKD1 and PKD2 genes encode for the integral membrane proteins polycystin-1 and polycystin-2, respectively, which are structurally similar and interact with one another.3,4 Mutations of PKD1 (85% of cases) or PKD2 (15% of cases) can lead to signal dysregulation and increased levels of cyclic adenosine monophosphate, culminating into cystogenesis.1,5,6 Given the dominant nature of transmission, there is at least a 50% probability that a child of an affected parent will inherit the disease. A spontaneous mutation causes ADPKD in 5% of cases.1
The progressive loss of nephrons secondary to cyst formation and expansion is not clinically detectable for the first few decades of life and is thought to be a result of compensatory hyperfiltration of intact renal tubules allowing maintenance of the glomerular filtration rate.7 Consequently, most patients may not develop notable renal insufficiency until their 30s or later, after which the glomerular filtration rate declines at an average of 4.4 to 5.9 mL per minute per year.8
Clinical Manifestations and Management
Many patients with ADPKD are asymptomatic, and only a few patients will present early with symptoms such as abdominal or flank pain, macroscopic hematuria, or urinary tract infections.9 The presenting features may be variable depending on the patient's comorbidities and can mimic other disease processes. Since the progression of ADPKD cannot be reversed with current treatment modalities, therapies target the resulting clinical manifestations.
CARDIOVASCULAR MANIFESTATIONS
Hypertension is the most common manifestation of ADPKD and contributes to renal dysfunction8 and cardiovascular complications, which are the most common causes of death in those with ADPKD.10 Patients with ADPKD and untreated hypertension are more likely to develop left ventricular hypertrophy earlier in the disease course. Left ventricular hypertrophy affects a significant proportion of children with ADPKD9,11; therefore, diagnosis and management of hypertension are vital. Angiotensin-converting enzyme inhibitors are recommended as first-line therapy, with angiotensin receptor blockers as the alternative for patients who cannot tolerate angiotensin-converting enzyme inhibitors.6,11–13 The goal of therapy should be blood pressure of less than 140/90 mm Hg in those who have developed chronic kidney disease; otherwise, the blood pressure targets are the same as those for the general population.13 Other common cardiac manifestations include mitral valve prolapse and aortic insufficiency secondary to aortic root dilatation.6,9
PAIN
Pain is the most common symptom in those with ADPKD and may present as back, chest, abdominal, or flank pain, or a combination.1,14 Although pain may be secondary to cyst rupture or the enlarged kidney compressing surrounding structures, it can also be due to a urinary tract infection or nephrolithiasis. Nonsteroidal anti-inflammatory drugs should be avoided because of renal toxicity.
Psychological assessment and depression screening should be considered for patients with chronic pain despite appropriate medical management. Adjuvant therapy for chronic pain includes antidepressants, lifestyle modification (e.g., avoidance of inciting activities, such as contact sports or heavy lifting), or reassurance. Referral to a pain management center may be necessary in patients at risk of narcotic dependence.6
URINARY TRACT INFECTIONS
Although symptomatic lower urinary tract infections are much more common in those with ADPKD than in the general population,15 the pattern of infections (more common in women) and the infectious organisms (gram-negative bacteria) are similar to those in the general population.16 Infected renal cysts usually yield urine cultures positive for Escherichia coli (74% of cases), with Staphylococcus aureus, Enterococcus, Lactobacillus, and anaerobic bacteria accounting for most of the remaining cases.17 However, negative urine culture results are possible when infected renal cysts are not in contact with the urinary space. Antibiotics with the ability to penetrate renal cysts include fluoroquinolones, third-generation cephalosporins (penetrate cysts poorly but with good parenchymal penetration), and trimethoprim/sulfamethoxazole.15,17–20 Fluoroquinolones are theoretically preferred because of their lipophilic properties, which allow for cyst diffusion, and bactericidal action against gram-negative pathogens.15,17
RENAL STONES
Nephrolithiasis is twice as common in those with ADPKD (lifetime prevalence of approximately 20%) than in the general population.1,21 Renal stones should be suspected in patients who have flank pain with or without hematuria. Fluid administration and pain management are first-line therapies.1,22 Extracorporeal shock wave lithotripsy and percutaneous nephrostolithotomy are also options.21
HEMATURIA
Gross hematuria occurs in more than 40% of persons with ADPKD23 and is a risk factor for the rapid progression of renal dysfunction, especially if it occurs before 30 years of age.24 The differential diagnosis includes abdominal trauma (e.g., from contact sports), urinary tract infections, renal stones, and cyst rupture into the urinary space. Hemorrhagic cysts can be diagnosed with computed tomography or magnetic resonance imaging and usually resolve within one week without treatment, although microscopic hematuria may still be present.1,22 If overt urinary bleeding lasts longer than one week or occurs for the first time in a patient older than 50 years, the patient should be evaluated for a neoplasm, which includes renal cell carcinoma or an unrelated cancer, such as bladder cancer.6,25,26
OTHER ABDOMINAL MANIFESTATIONS
Although extrarenal cysts can appear in the seminal vesicles, ovaries, pancreas, spleen, and central nervous system, hepatic cysts are the most common.9,27,28 Other than a mild rise in serum alkaline phosphatase levels, the patient is typically asymptomatic and has preserved liver function. However, symptoms resulting from significant liver enlargement can include shortness of breath, early satiety or loss of appetite, abdominal discomfort, esophageal reflux, lower extremity edema, low back pain, and rarely inferior vena cava compression.6,9 Acute complications include infection or hemorrhage of hepatic cysts.27 Other abdominal manifestations of ADPKD include diverticulosis, and abdominal and inguinal hernias.
Because of the proliferative effect of estrogen on hepatic cysts, rapid and significant hepatic cyst enlargement is more likely during pregnancy and with estrogen therapy.9 Consequently, oral contraceptives containing estrogen and menopausal estrogen therapy should be administered at the lowest effective dose or avoided in patients with ADPKD.1,6,27
INTRACRANIAL ANEURYSM
Intracranial aneurysm represents the most severe extra-renal manifestation in those with ADPKD.9 Although the prevalence of intracranial aneurysm in patients with ADPKD varies, it is at least twice as likely to occur in these patients than in the general population; the prevalence is even higher in patients with ADPKD who also have a family history of intracranial aneurysms.29–31 The complications of intracranial aneurysms include thromboembolism, localized intracranial pressure with mass effect, and fatal subarachnoid hemorrhage,29,32 although most aneurysms less than 7 mm in diameter and in the anterior circulation are less likely to rupture and can be monitored.33 The use of time-of-flight magnetic resonance angiography (i.e., inflow angiography, which does not require contrast) is recommended to screen for aneurysms29 when indicated (Table 1).29,31,32
Table 1. Indications to Screen for Intracranial Aneurysm in Patients with ADPKD

| Before major elective surgery |
| Central nervous system signs or symptoms (e.g., nausea and vomiting, lethargy, photophobia, focal signs, seizure, transient ischemic attack, loss of consciousness) |
| Family history of intracranial aneurysm or intracranial hemorrhage |
| High-risk occupation (e.g., airline pilot) |
| New-onset severe headache |
ADPKD = autosomal dominant polycystic kidney disease.
ADPKD AND PREGNANCY
Fertility rates for both men and women with ADPKD who have not developed significant renal dysfunction are comparable to those of the general population. However, women with ADPKD and hypertension are more likely to develop fetal complications (32.6% vs. 26.2%) and maternal complications (35% vs. 19%) during pregnancy than women with ADPKD who are normotensive.34
END-STAGE RENAL DISEASE
All patients with ADPKD develop a progressive loss of renal function, with nearly 80% of patients dying or developing end-stage renal disease by 70 years of age.25 However, those with the PKD1 mutation will develop end-stage renal disease approximately 10 to 15 years before those with the PKD2 mutation.9 If renal replacement therapy is required, peritoneal dialysis and hemodialysis are options. The overall survival rate of renal transplant recipients who develop end-stage renal disease from ADPKD is comparable to that of renal transplant recipients who have end-stage renal disease from other causes.35
Screening
Patients with a family history of ADPKD are candidates for screening, typically with imaging studies.6 Since no interventions are known to prevent cyst development, screening should be delayed until at least 18 years of age.1,6 If ADPKD is diagnosed, patients should be encouraged to inform their first-degree relatives so that they have the opportunity to be screened as well.1
DIAGNOSTIC IMAGING
Although computed tomography and magnetic resonance imaging are slightly more sensitive for detecting renal cysts, ultrasonography is the preferred screening modality because of lower cost, increased availability, and lack of radiation exposure.9,36 Computed tomography and magnetic resonance imaging may have a role in screening morbidly obese patients with nondiagnostic ultrasound results.36 Diagnostic criteria for ADPKD are listed in Table 2.36–38
Table 2. Radiographic Diagnostic Criteria for ADPKD

| Age (years) | Number of cysts | |
|---|---|---|
| Ultrasonography (at-risk of ADPKD type 1) | ||
| < 30 | ≥ 2 in one or both kidneys | |
| 30 to 59 | ≥ 2 in each kidney | |
| ≥ 60 | ≥ 4 in each kidney | |
| Ultrasonography (at risk and unknown genotype) | ||
| 15 to 39 | ≥ 3 in one or both kidneys | |
| 40 to 59 | ≥ 2 in each kidney | |
| ≥ 60 | ≥ 4 in each kidney | |
| Magnetic resonance imaging (at risk) | ||
| < 30 | ≥ 5 in each kidney | |
| 30 to 44 | ≥ 6 in each kidney | |
| 45 to 59 (females) | > 6 in each kidney | |
| 45 to 59 (males) | > 9 in each kidney | |
ADPKD = autosomal dominant polycystic kidney disease.
GENETIC TESTING
Genetic testing is not standard practice in the diagnosis of ADPKD, but it is useful in certain scenarios, such as in a young at-risk patient who is asymptomatic and has negative findings on renal ultrasonography; in a patient with an unknown family history and a serendipitous finding of enlarged kidneys with renal cysts; and in an at-risk patient who wishes to be a kidney transplant donor.6,39 It is also a confirmatory test that does not rely on age-specific criteria. Genetic testing, when indicated, can be completed via linkage or sequence analysis.6
Data Sources: We searched PubMed using the key terms ADPKD or autosomal dominant polycystic kidney disease. The search included meta-analyses, randomized controlled trials, clinical trials, practice guidelines, genetics, symptoms, therapy, and reviews. We also searched Essential Evidence Plus, the National Guideline Clearinghouse, Agency for Healthcare Research and Quality Evidence Reports, U.S. Preventive Services Task Force guidelines, the National Institute for Health and Clinical Excellence, and the Cochrane Database of Systematic Reviews. Search dates: September 2012, March to August 2013, and July 2014.
