Pulmonary hypertension is a common, complex group of disorders that result from different pathophysiologic mechanisms but are all defined by a mean pulmonary arterial pressure of 25 mm Hg or greater. Patients often initially present to family physicians; however, because the symptoms are typically nonspecific or easily attributable to comorbid conditions, diagnosis can be challenging and requires a stepwise evaluation. There is limited evidence to support screening of asymptomatic individuals. Echocardiography is recommended as the initial step in the evaluation of patients with suspected pulmonary hypertension. A definitive diagnosis cannot be made on echocardiographic abnormalities alone, and some patients require invasive evaluation by right heart catheterization. For certain categories of pulmonary hypertension, particularly pulmonary arterial hypertension, treatment options are rapidly evolving, and early diagnosis and prompt referral to an expert center are critical to ensure the best prognosis. There are no directed therapies for many other categories of pulmonary hypertension; therefore, family physicians have a central role in managing contributing comorbidities. Other important considerations for patients with pulmonary hypertension include influenza and pneumonia immunizations, contraception counseling, preoperative assessment, and mental health.
Pulmonary hypertension is a heterogeneous group of disorders characterized by a mean pulmonary arterial pressure of 25 mm Hg or greater at rest during right heart catheterization.1–4 Patients with pulmonary hypertension may initially present to family physicians, but symptoms such as dyspnea on exertion and fatigue are nonspecific and may be attributed to comorbid conditions.5 Pulmonary hypertension is categorized on the basis of pathophysiology, hemodynamics, and therapeutic options. In 2013, the classification scheme was updated to recognize five groups of pulmonary hypertension6 (Table 13,4,6 ).
SORT: KEY RECOMMENDATIONS FOR PRACTICE

| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Echocardiography is the recommended initial test in the evaluation of patients with suspected pulmonary hypertension. | C | 1–3, 22 |
| Results from invasive hemodynamic testing with right heart catheterization, preferably at an expert center, should be obtained before initiating treatment with vasodilator therapy in patients with pulmonary arterial hypertension. | C | 1–3 |
| In patients with pulmonary hypertension due to lung disease or left heart disease, treatment should focus on optimizing comorbid conditions. | C | 3, 9–11 |
| In patients with pulmonary hypertension and hypoxia, supplemental oxygen should be administered to maintain saturation above 90%. | C | 11, 28, 29 |
| Use of vasodilator therapies in patients with pulmonary hypertension due to lung disease or left heart disease is potentially harmful and not recommended. | C | 10, 11, 26 |
| Patients with chronic thromboembolic pulmonary hypertension should receive lifelong anticoagulation in the absence of contraindications. | C | 25 |
| Patients with pulmonary hypertension should receive seasonal influenza vaccination and age-appropriate pneumococcal vaccination, unless contraindicated. | A | 1, 31, 32 |
| Perioperative assessment of patients with pulmonary hypertension should include echocardiographic assessment of right ventricular function. | C | 33 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
Table 1. Clinical Classification of Pulmonary Hypertension

| Classification | Targeted treatment available? |
|---|---|
| Group 1*: Pulmonary arterial hypertension | Yes |
| Including idiopathic, heritable, and HIV-associated; systemic sclerosis and other connective tissue disease; congenital heart disease; schistosomiasis; drug- and toxin-induced | |
| Group 2: Pulmonary hypertension due to left heart disease | No |
| Including systolic and diastolic dysfunction and valvular heart disease | |
| Group 3: Pulmonary hypertension due to lung diseases and/or hypoxia | No |
| Including chronic obstructive pulmonary disease, sleep-disordered breathing, and interstitial lung disease | |
| Group 4: Chronic thromboembolic pulmonary hypertension | Yes |
| Group 5: Multifactorial pulmonary hypertension | No |
| Including metabolic, systemic, and hematologic disorders (sickle cell disease), and others |
HIV = human immunodeficiency virus.
*—Also includes 1′ (pulmonary venoocclusive disease and/or pulmonary capillary hemangiomatosis) and 1″ (persistent pulmonary hypertension of the newborn).
Although pulmonary arterial hypertension (group 1) has benefited the most from progress with targeted therapy, this diagnosis is rare and may not be encountered by many family physicians.7 However, in certain populations, such as persons with systemic sclerosis, pulmonary arterial hypertension is more common.8 Treatment is rapidly evolving and usually administered in expert centers. The American College of Chest Physicians published treatment guidelines in 2014.1
Family physicians most often encounter pulmonary hypertension in patients with chronic disease such as heart failure, obstructive lung disease, and thromboembolism. Pulmonary hypertension is most prevalent in those with left heart disease (group 2).9 In systolic or diastolic left heart failure, prevalence estimates range from 25% to 83%.10 Pulmonary hypertension is also common in patients with lung disease and/or hypoxia (group 3). In patients with chronic obstructive pulmonary disease (COPD), the prevalence of pulmonary hypertension increases with COPD severity. It is found in 20% of patients who have been hospitalized and in more than 50% of patients with end-stage disease.11 After an acute pulmonary embolism, one study found a 3.8% incidence of chronic thromboembolic pulmonary hypertension (group 4) after two years.12 Group 5 is multifactorial in etiology and best characterized in the setting of sickle cell disease, where it is an independent predictor of mortality.13,14 The prevalence of pulmonary hypertension increases with age. A population-based study in Minnesota estimated that the total prevalence could reach 10% to 20% in the general population.15
There is limited evidence to guide screening for pulmonary hypertension in asymptomatic individuals, even in high-risk groups, which leads to significant delays in diagnosis. A multicenter retrospective study of patients with idiopathic pulmonary arterial hypertension found a mean time from symptom onset to diagnosis of 47 (± 34) months encompassing 5.3 (± 3.8) primary care visits and evaluation by 3.0 (± 2.1) subspecialists.16 Registries from the United Kingdom and Ireland show that in patients older than 50 years, the median duration of symptoms at diagnosis was 24 months vs. 12 months for patients younger than 50 years. This likely reflects the challenges of diagnosis in patients with comorbidities.5 Reducing this delay allows initiation of therapy before right heart failure develops.1
Independent of classification, pulmonary hypertension can cause progressive, disabling symptoms, as well as increases in morbidity and mortality. For example, pulmonary hypertension status in patients who also have COPD may be more predictive of mortality than pulmonary function markers, such as forced expiratory volume in one second or diffusing capacity.11 Patients with pulmonary arterial hypertension have a 36% five-year mortality rate.17
Pathophysiology
The mechanisms that increase pulmonary pressures can act primarily on the pulmonary arterial bed or venous bed, either alone or in combination.3,18 Pulmonary arterial hypertension is characterized by progressive narrowing of distal pulmonary arteries attributed to a variety of pathologic insults, such as arterial vasoconstriction, medial hypertrophy, intimal proliferation, and fibrosis.19 There are some genetic associations, such as BMPR2, but these are insufficient to explain the pathogenesis without other contributing factors.20 Pulmonary hypertension due to left heart disease is primarily a pulmonary venous process and likely results from passive pulmonary venous congestion with vasoconstriction and venous remodeling.3,9 In pulmonary hypertension due to lung disease and/or hypoxia, increases in pulmonary arterial pressures may arise from destruction of the alveolar capillary bed or chronic hypoxic vasoconstriction.3 Chronic thromboembolic pulmonary hypertension develops following thrombotic macrovascular obstruction with subsequent vasoconstriction and remodeling of the pulmonary arterial bed.19
Regardless of the mechanism, persistently increased pulmonary arterial pressure strains the thin-walled right ventricle. Adapted to the low-pressure pulmonary circulation, the right ventricle is unable to sustain cardiac output with these pressures.3 Ultimately, right ventricular failure is the most common cause of death in patients with pulmonary hypertension.21
Screening and Diagnostic Evaluation
There are no data to support screening of asymptomatic individuals for pulmonary hypertension, even in high-risk groups such as those with a family history of pulmonary arterial hypertension without known BMPR2 mutations.3,22 For patients with systemic sclerosis and related diseases, expert opinion suggests annual screening with laboratory testing (including brain natriuretic peptide), electrocardiography, and pulmonary function testing, followed by echocardiography.3,8 Expert consensus recommends annual echocardiography in patients with sickle cell disease and those with known BMPR2 mutations.3,14
Recognizing pulmonary hypertension in patients presenting with new signs or symptoms can be difficult because many symptoms are common and associated with an extensive differential diagnosis (Table 21–3,22 ). Pulmonary hypertension should be considered in patients with chronic illness and symptoms that are disproportionate to the underlying disease or poorly responsive to treatment.
Table 2. Findings Suggestive of Pulmonary Hypertension

| History |
| Angina |
| Dyspnea on exertion |
| Exercise intolerance |
| Fatigue |
| History of medical illness associated with pulmonary hypertension |
| Syncope or presyncope |
| Physical examination |
| Abnormal pulse oximetry, elevated jugular venous pressure, ascites, lower extremity edema, right ventricular heave, tricuspid regurgitation murmur (increased pulmonic component of S2) |
| Signs of right heart failure |
| Office-based diagnostic tests |
| Chest radiography: right ventricular enlargement, engorged pulmonary arteries |
| Electrocardiography: right ventricular enlargement, right bundle branch block, right ventricular strain pattern, S1Q3T3 pattern |
| Laboratory studies: elevated brain natriuretic peptide level |
A stepwise approach can minimize the risks and costs of unnecessary testing (Figure 1). Physicians should initially consider the history, clinical findings, and targeted noninvasive testing, particularly echocardiography. Patients presenting with signs of advanced pulmonary hypertension (right heart failure or syncope) should be promptly evaluated with echocardiography. Some patients require right heart catheterization, which is important for classification and subsequent treatment options.1 However, not all patients with evidence of pulmonary hypertension on noninvasive testing need catheterization. For example, those with mildly increased systolic pulmonary arterial pressure (i.e., less than 35 mm Hg) and an established precipitating diagnosis often do not need catheterization.
Figure 1. Evaluation of Suspected Pulmonary Hypertension in Primary Care
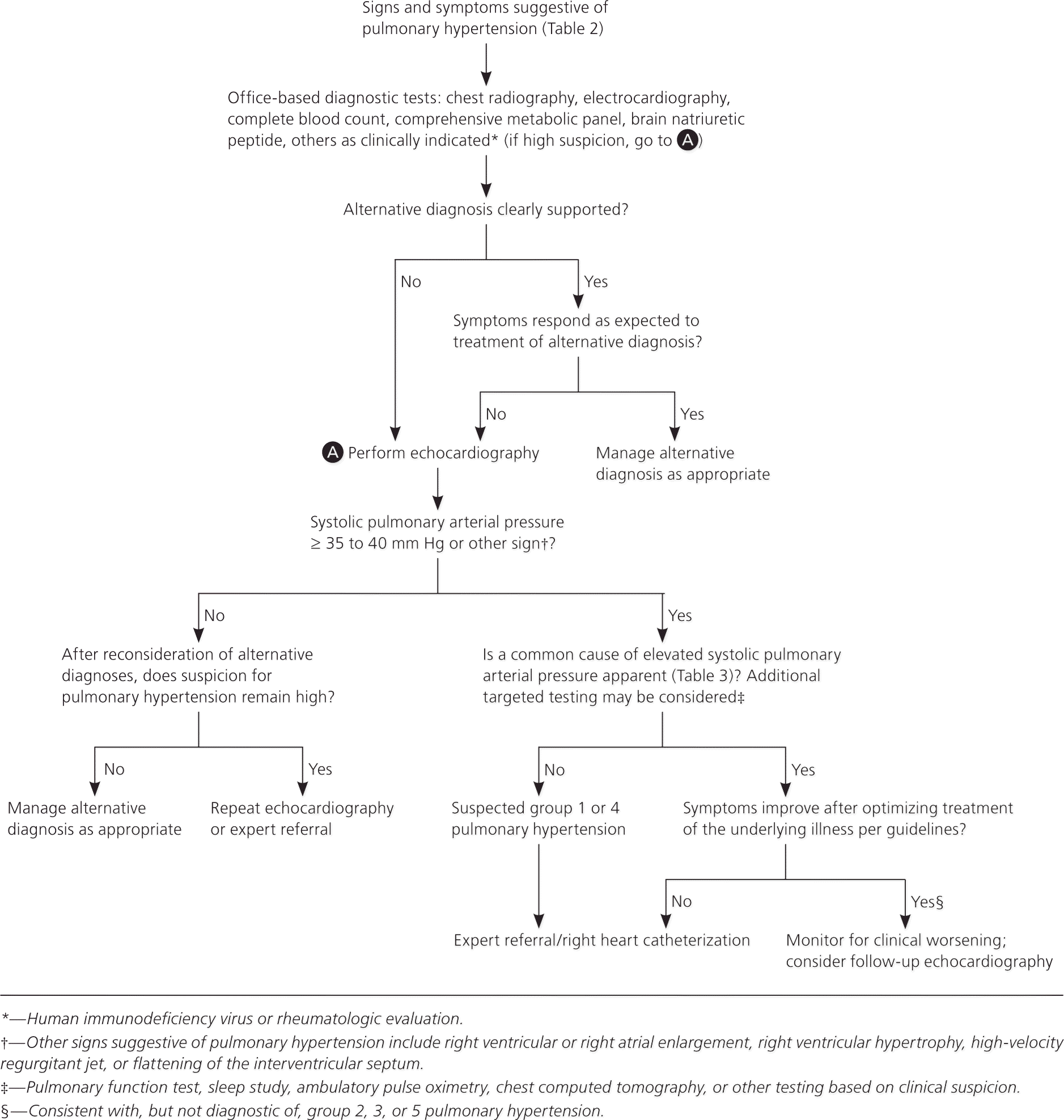
Suggested algorithm for the evaluation of suspected pulmonary hypertension in primary care.
Echocardiography is the initial noninvasive diagnostic test, according to national guidelines.1–3,22 It is readily available and provides information about abnormalities in undiagnosed patients. An estimated systolic pulmonary arterial pressure of 35 to 40 mm Hg or greater on echocardiography is suggestive of pulmonary hypertension.1,21,22 Echocardiography also assesses right heart function, which is needed for diagnosis and monitoring disease progression.21
A meta-analysis calculated the accuracy of echocardiography vs. right heart catheterization for diagnosing pulmonary hypertension and found a sensitivity of 83% (95% confidence interval [CI], 73 to 90), a specificity of 72% (95% CI, 53 to 85), and an odds ratio of 13 (95% CI, 5 to 31).23 Elevated systolic pulmonary arterial pressure occurs in other conditions (Table 321–23 ), and echocardiography and right heart catheterization results can be disparate. In an observational U.S. cohort study, approximately 60% of echocardiography estimates of systolic pulmonary arterial pressure had a difference of more than 10 mm Hg vs. catheterization.24 On meta-analysis, the correlation between systolic pulmonary arterial pressure on echocardiography compared with heart catheterization was 0.70 (95% CI, 0.67 to 0.73).23
Table 3. Common Causes of Elevated Systolic Pulmonary Arterial Pressure*

| Chronic lung disease and sleep disorders (including chronic obstructive pulmonary disease and obstructive sleep apnea) |
| High cardiac output states (e.g., anemia, hyperthyroidism) |
| Hypertension |
| Left heart disease, including heart failure with preserved or reduced ejection fraction |
| Obesity |
| Volume overload, particularly in heart failure or in the setting of dialysis and chronic kidney disease |
*—Other than increased vascular resistance.
Additional testing, including right heart catheterization, depends on the differential and the treatment response.3 For example, if chronic thromboembolic pulmonary hypertension is suspected, a ventilation-perfusion scan is part of the initial evaluation, but in a patient with underlying chronic lung disease, pulmonary function tests should be performed.3,25 Subspecialty consultation may be useful, particularly for patients with symptoms that progress despite treatment of comorbidities.3
Treatment
The diagnostic classification of pulmonary hypertension determines the treatment options. Evidence supporting targeted treatment is available only for pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension.1,22 For the other classes, there are no data on treatment effectiveness, and for some therapies, there is evidence of harm. For example, in the Choosing Wisely campaign, treatment with advanced vasoactive agents is recommended only for the management of pulmonary arterial hypertension.26
PULMONARY ARTERIAL HYPERTENSION
Drug development has focused on the treatment of patients with pulmonary arterial hypertension. Studies are limited by short follow-up periods and a lack of patient-centered outcomes.1,22 Patients should have a right heart catheterization and subspecialty referral before initiation of vasodilator or other targeted therapies.1–3 Patients without symptoms or evidence of functional impairment (using a six-minute walk test) should generally be monitored without therapy.1 After symptoms develop, patients with acute vasoreactivity on right heart catheterization should begin a trial of calcium channel blockers.1 Patients with a mean pulmonary arterial pressure decrease of more than 10 mm Hg to less than 40 mm Hg and with an unchanged or increased cardiac output when challenged are considered vasoreactive.3 Other treatments may include an endothelin receptor antagonist (bosentan [Tracleer]), a phosphodiesterase type 5 inhibitor (sildenafil [Revatio]), or a soluble guanylate cyclase stimulator (riociguat [Adempas]).1 Further treatment may include parenteral or inhaled prostanoids, such as epoprostenol (Flolan, Veletri), and newer oral prostacyclin agents, such as selexipag (Uptravi) and treprostinil (Orenitram).1,27
PULMONARY HYPERTENSION DUE TO LEFT HEART DISEASE
The focus for these patients is optimizing the underlying heart disease and controlling comorbidities.3,9,10 This includes management of hypertension and heart failure, and addressing significant valvular disease when present. Control of fluid volume and diuretic therapy are essential, particularly in patients with a history of volume overload or right heart failure.10 Vasodilators are not recommended for the treatment of pulmonary hypertension due to left heart disease.10,26
PULMONARY HYPERTENSION DUE TO LUNG DISEASE
The treatment of pulmonary hypertension due to lung disease should focus on managing the underlying lung disease and optimizing treatment of other comorbidities.3,11 Lung disease should be treated according to the best available evidence.28,29 Patients with COPD and arterial oxygen pressures less than 60 mm Hg should receive supplemental oxygen, which may improve mortality by lowering pulmonary arterial pressures.11,29 Patients should be screened for obstructive sleep apnea and treated when necessary.2,30 Patients with hypoxic lung disease may benefit from supplemental oxygen to maintain saturation greater than 90%.11 The use of vasodilators in chronic lung disease may worsen ventilation-perfusion mismatching.11,26 Patients with chronic lung disease and severe pulmonary hypertension should consult with a subspecialist.11
CHRONIC THROMBOEMBOLIC PULMONARY HYPERTENSION
A pulmonary endarterectomy performed at a center of excellence can be curative and is first-line therapy in patients with chronic thromboembolic pulmonary hypertension who are surgical candidates. Patients who are not surgical candidates can be considered for targeted medical therapies also used to treat pulmonary arterial hypertension. Regardless of the treatment, these patients should receive lifelong anticoagulation therapy in the absence of contraindications.25
MULTIFACTORIAL PULMONARY HYPERTENSION
There are limited data on the treatment of pulmonary hypertension for most of the etiologies in this group. In patients with sickle cell disease, guidelines recommend initiating hydroxyurea in patients with elevated mortality risk. Chronic transfusion therapy is a second-line option. The use of therapies targeted at pulmonary arterial hypertension is strongly discouraged.14
Other Considerations
IMMUNIZATION
Patients with pulmonary hypertension should have seasonal influenza vaccination. Age-based recommendations for pneumococcal vaccination should be followed, including use of the 23-valent pneumococcal polysaccharide vaccine (Pneumovax) for adults younger than 65 years.1,31,32
PERIOPERATIVE ASSESSMENT
Pulmonary hypertension is associated with increased morbidity and mortality during the perioperative period.1,33 The perioperative assessment should include an assessment of functional status, care goals, and alternatives to surgery.33 Echocardiography assessing right ventricular function can help determine surgical risk.33 Surgical management of patients with severe pulmonary hypertension should be performed with subspecialty consultation.1,33
CONTRACEPTION
There is an elevated risk of pregnancy complications in women with pulmonary hypertension and particularly pulmonary arterial hypertension. Current guidelines discourage pregnancy and recommend contraception counseling with emphasis on prescribing a long-acting, highly effective method of contraception.34
MENTAL HEALTH AND PATIENT EDUCATION
An ethnographic study of patients with pulmonary hypertension found that many described uncertainty surrounding their prognosis and expressed feelings of isolation.35 Patients may benefit from resources available through the Pulmonary Hypertension Association (http://www.phassociation.org).
Data Sources: A PubMed search was completed using the terms pulmonary hypertension and pulmonary arterial hypertension. We also searched the National Guideline Clearinghouse. Our search included consensus guidelines, systematic reviews, meta-analyses, randomized controlled trials, and large retrospective or cohort studies. Search dates: May to December 2015.
