Each year, 4 to 5 million newborns receive state-mandated screening. Although the Advisory Committee on Heritable Disorders in Newborns and Children has identified 34 core conditions that should be incorporated into screening programs, each state manages, funds, and maintains its own program. State programs encompass screening, as well as the diagnosis and coordination of care for newborns with positive findings. Testing for core disorders is fairly standardized, but more extensive screening varies widely by state, and the rigorous evaluation of new screening panels is ongoing. The core panel includes testing for three main categories of disorders: metabolic disorders (e.g., amino acid and urea cycle, fatty acid oxidation, and organic acid disorders); hemoglobinopathies; and a group of assorted conditions, including congenital hearing loss. Family physicians must be familiar with the expanded newborn screening tests to effectively communicate results to parents and formulate interventions. They must also recognize signs of metabolic disorders that may not be detected by screening tests or that may not be a part of standard newborn screening in their state. For infants with positive screening results leading to diagnosis, long-term follow-up involves ongoing parental education, regular medical examinations, management at a metabolic treatment center, and developmental and neuropsychological testing to detect associated disorders in time for early intervention.
Newborn screening programs are state-mandated public health services aimed at ensuring that the 4 to 5 million infants born each year are screened for certain serious conditions at birth, allowing for treatment before harmful effects can occur.1,2 Although these conditions are rare, they are detected on screening in 5,000 to 6,000 newborns every year. The combined incidence of screened conditions is estimated to be as high as one per 500 to 1,000 births.3 If they are not detected and treated early, these conditions can lead to adverse outcomes, including moderate to severe neuropsychological dysfunction, intellectual developmental disabilities, and death.
WHAT IS NEW ON THIS TOPIC: NEWBORN SCREENING
The Advisory Committee on Heritable Disorders in Newborns and Children recommends that every newborn screening program include a Recommended Uniform Screening Panel that screens for 34 core disorders and 26 secondary disorders.
The American College of Medical Genetics and Genomics has published action sheets and algorithms for physicians to use if a newborn screening result is abnormal. Detailed fact sheets on many disorders have been published in Pediatrics and are available free of charge on the journal's website.
SORT: KEY RECOMMENDATIONS FOR PRACTICE
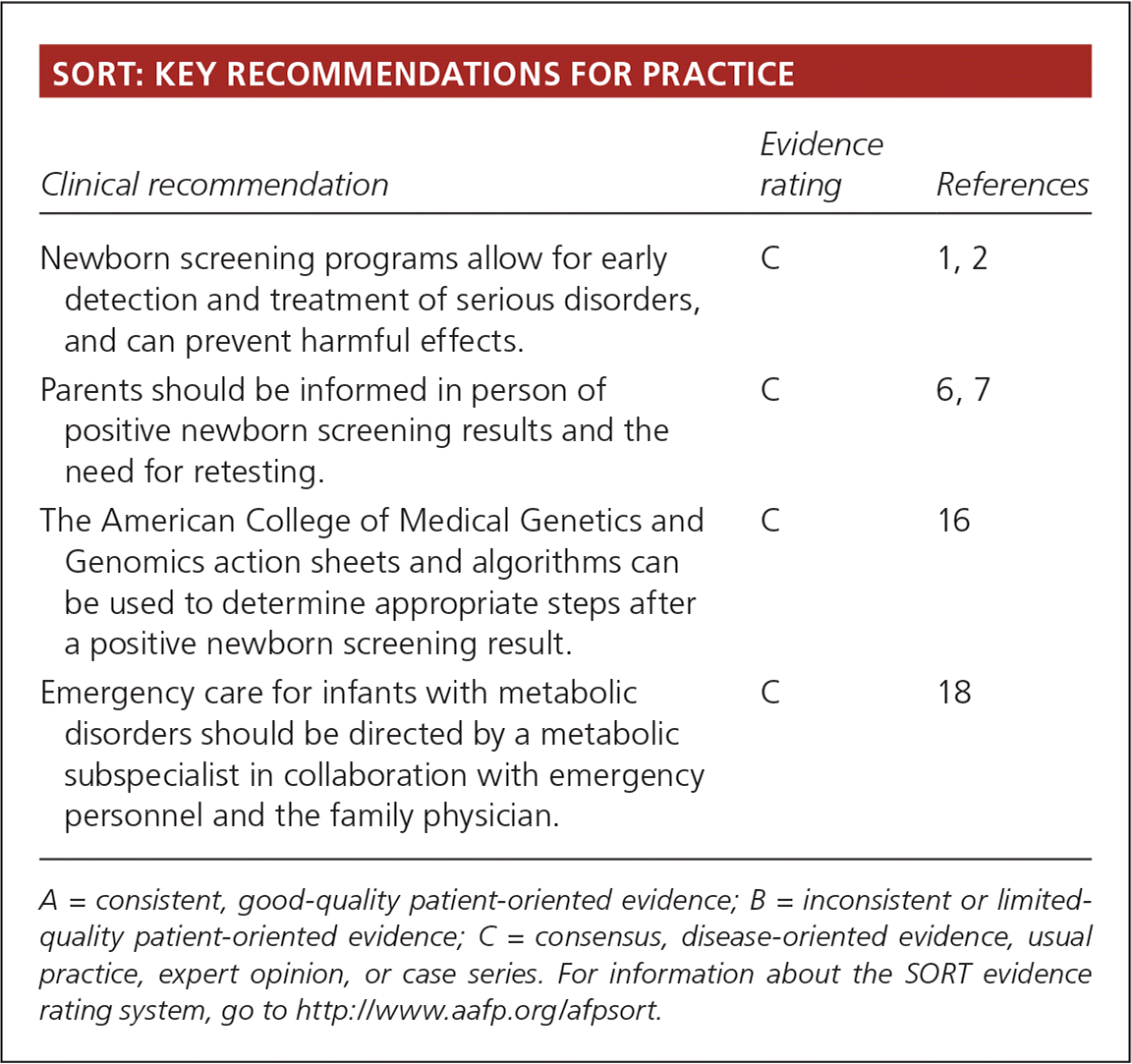
| Clinical recommendation | Evidence rating | References |
|---|---|---|
| Newborn screening programs allow for early detection and treatment of serious disorders, and can prevent harmful effects. | C | 1, 2 |
| Parents should be informed in person of positive newborn screening results and the need for retesting. | C | 6, 7 |
| The American College of Medical Genetics and Genomics action sheets and algorithms can be used to determine appropriate steps after a positive newborn screening result. | C | 16 |
| Emergency care for infants with metabolic disorders should be directed by a metabolic subspecialist in collaboration with emergency personnel and the family physician. | C | 18 |
A = consistent, good-quality patient-oriented evidence; B = inconsistent or limited-quality patient-oriented evidence; C = consensus, disease-oriented evidence, usual practice, expert opinion, or case series. For information about the SORT evidence rating system, go to https://www.aafp.org/afpsort.
To encourage uniform and comprehensive newborn screening across the states, the Advisory Committee on Heritable Disorders in Newborns and Children was chartered in 2015. The committee advises the Secretary of Health and Human Services on the most appropriate application of newborn screening tests, technologies, policies, guidelines, and standards. The committee recommends that every newborn screening program include the Recommended Uniform Screening Panel, which currently screens for 34 core disorders and 26 secondary disorders.1,2 However, all states have laws mandating newborn screening, with defined opt-out policies for parents,4 and each state independently determines which screening tests are performed and what follow-up is provided. All but four state programs are at least partially fee based, with an average initial newborn screening fee of approximately $76 in 2015.5 For more information on state-specific programs, go to http://genes-r-us.uthscsa.edu/resources/consumer/statemap.htm.
The first newborn screening test in the United States was developed more than 50 years ago to detect phenylketonuria, a biochemical genetic disorder. Developments in technology and genomic medicine have increased the scope of newborn screening, and now there are screening tests for more than 60 conditions. Tandem mass spectrometry (MS/MS), which allows for screening of many disorders using a single blood sample, is used in all states. Real-time polymerase chain reactions, electrophoresis, and enzyme assays also use a dried blood spot. The newborn blood sample is obtained at 24 to 48 hours of life, and screening results are generally available within 24 hours.
Local and regional metabolic subspecialists and treatment centers familiar with specific disorders (see http://genes-r-us.uthscsa.edu/resources.htm) assist primary care physicians when screening results are positive. This includes clearly communicating follow-up testing recommendations and the degree of severity and suspicion to aid the physician when discussing the disorder with the family. Parents should be informed in person about the positive results and the need for retesting,6,7 and should be given an explanation of what the initial and follow-up findings mean, even if the diagnosis is not confirmed.8
Disorders Included in Newborn Screening Programs
The core tests in the Recommended Uniform Screening Panel include three main categories of disorders: metabolic disorders; hemoglobinopathies; and a group of assorted conditions, including congenital hearing loss.9
METABOLIC DISORDERS
Amino acid and urea cycle, fatty acid oxidation, and organic acid disorders can be detected with MS/MS. Despite the high sensitivity and specificity of MS/MS screening, the low prevalence of screened diseases leads to an increased number of false-positive results. Most screening tests for metabolic disorders are normal. The next most common result is a borderline result, indicating a value above the upper limit of normal but below the lower limit of abnormal typically required for the diagnosis of a disorder. In many cases, a repeat sample will be in the normal range, and parents can be assured that the screening is negative.10 Upper limits of normal are used to avoid missing inborn errors of metabolism that have not yet presented because the infant has not been exposed to sufficient quantities of the precursors of toxic metabolites.
Positive screening results require urgent attention. Sometimes, a markedly abnormal result necessitates immediate intervention, including hospitalization or examination by a metabolic subspecialist or clinical geneticist. If there is uncertainty regarding results, a metabolic subspecialist should be consulted. In most states, the newborn screening program maintains contact information for subspecialists who are on call (see http://genes-r-us.uthscsa.edu/resources.htm and http://www.babysfirsttest.org/newborn-screening/states).
Amino acid disorders (Table 13,11–13 ) are treated with a protein-restricted diet. Infant formula that provides the necessary parts of protein but not the offending amino acid is available. Despite treatment, children may develop learning disabilities, attention-deficit/hyperactivity disorder, and emotional problems. Urea cycle disorders are more recent additions to newborn screening that require urgent action. These disorders can result in severe cognitive deficits, illness, and death, and patients should be referred to a multidisciplinary team at a metabolic center. Infants with metabolic disorders associated with acute neonatal illness may exhibit neurobehavioral symptoms, including lethargy, poor feeding, and vomiting. Caregivers will often note that the child “doesn't look right.” Such observations require immediate action within the context of an abnormal newborn screening result.
Table 1. Newborn Screening: Amino Acid Disorders
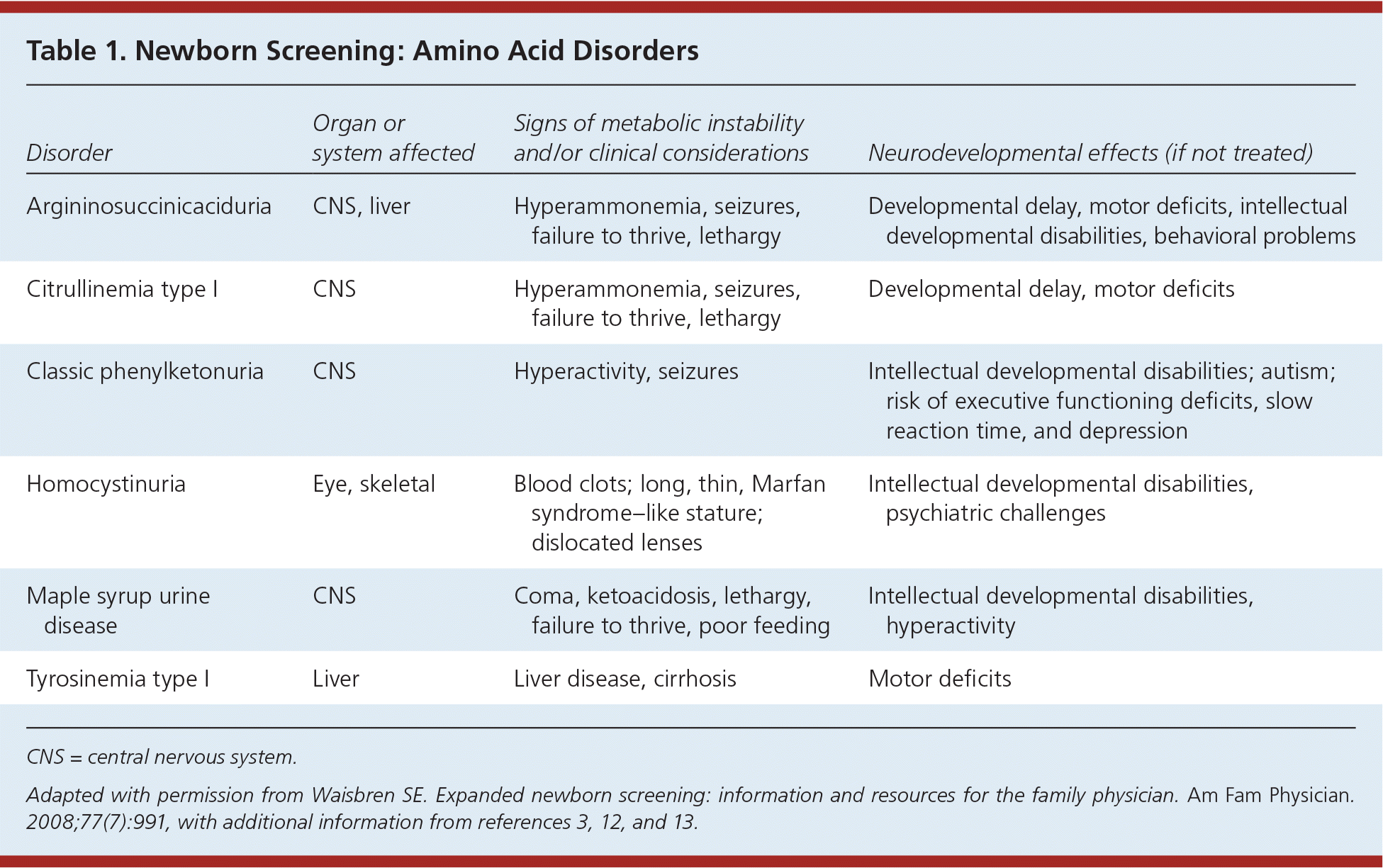
| Disorder | Organ or system affected | Signs of metabolic instability and/or clinical considerations | Neurodevelopmental effects (if not treated) |
|---|---|---|---|
| Argininosuccinicaciduria | CNS, liver | Hyperammonemia, seizures, failure to thrive, lethargy | Developmental delay, motor deficits, intellectual developmental disabilities, behavioral problems |
| Citrullinemia type I | CNS | Hyperammonemia, seizures, failure to thrive, lethargy | Developmental delay, motor deficits |
| Classic phenylketonuria | CNS | Hyperactivity, seizures | Intellectual developmental disabilities; autism; risk of executive functioning deficits, slow reaction time, and depression |
| Homocystinuria | Eye, skeletal | Blood clots; long, thin, Marfan syndrome–like stature; dislocated lenses | Intellectual developmental disabilities, psychiatric challenges |
| Maple syrup urine disease | CNS | Coma, ketoacidosis, lethargy, failure to thrive, poor feeding | Intellectual developmental disabilities, hyperactivity |
| Tyrosinemia type I | Liver | Liver disease, cirrhosis | Motor deficits |
CNS = central nervous system.
Adapted with permission from Waisbren SE. Expanded newborn screening: information and resources for the family physician. Am Fam Physician . 2008;77(7):991, with additional information from references 3, 12, and 13.
Fatty acid oxidation disorders (Table 23,11–14 ) are associated with central nervous system, cardiovascular, and other clinical anomalies, as well as intellectual developmental disabilities.14 Treatment generally consists of avoidance of fasting. Most infants without metabolic episodes develop normal cognitive abilities. Medium-chain acyl-CoA dehydrogenase deficiency is the most common fatty acid oxidation disorder and may be associated with intermittent severe metabolic crisis or sudden death.15 The disorder is almost completely asymptomatic until the child has an acute fasting illness.
Table 2. Newborn Screening: Fatty Acid Oxidation Disorders
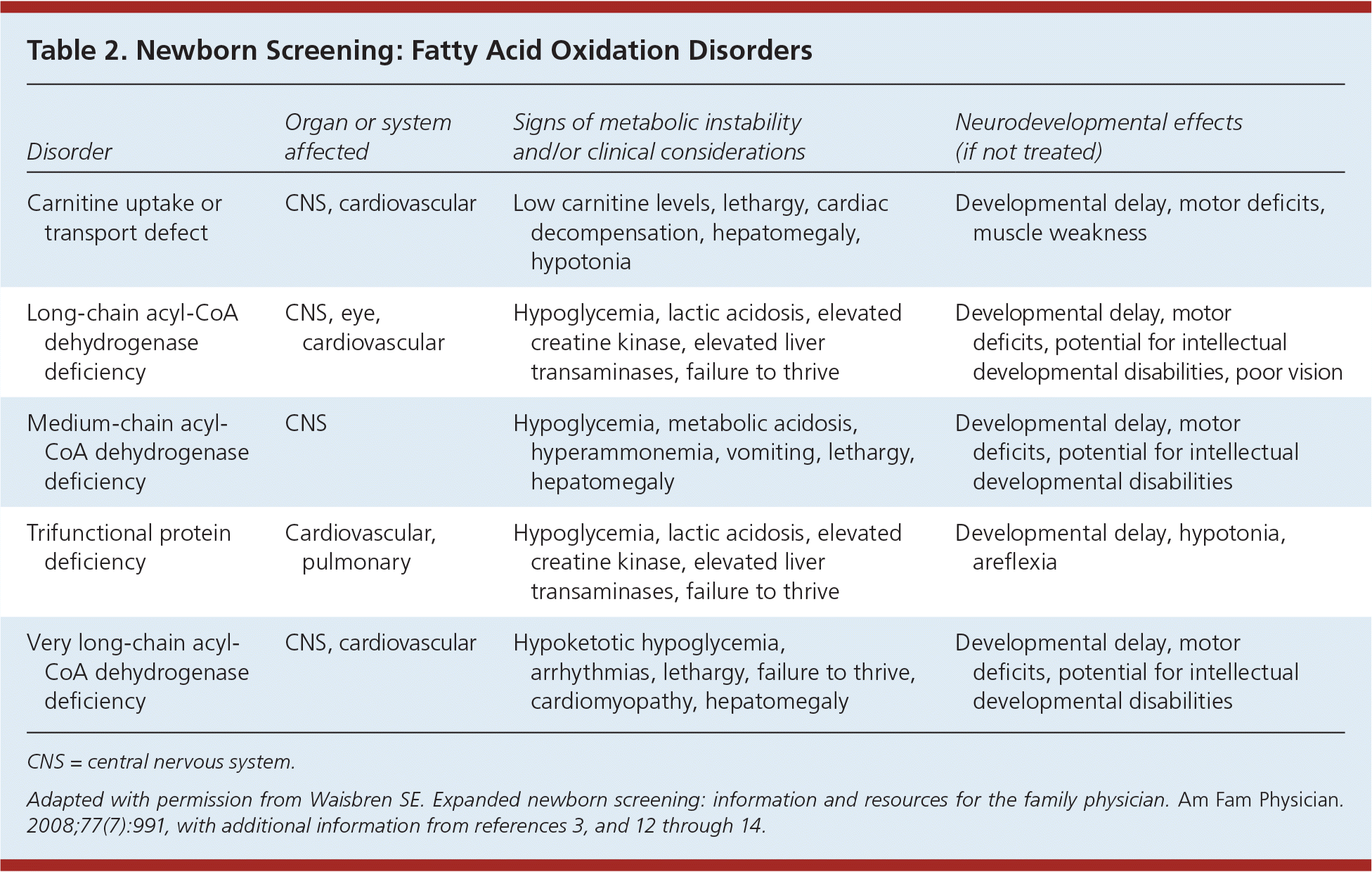
| Disorder | Organ or system affected | Signs of metabolic instability and/or clinical considerations | Neurodevelopmental effects (if not treated) |
|---|---|---|---|
| Carnitine uptake or transport defect | CNS, cardiovascular | Low carnitine levels, lethargy, cardiac decompensation, hepatomegaly, hypotonia | Developmental delay, motor deficits, muscle weakness |
| Long-chain acyl-CoA dehydrogenase deficiency | CNS, eye, cardiovascular | Hypoglycemia, lactic acidosis, elevated creatine kinase, elevated liver transaminases, failure to thrive | Developmental delay, motor deficits, potential for intellectual developmental disabilities, poor vision |
| Medium-chain acyl-CoA dehydrogenase deficiency | CNS | Hypoglycemia, metabolic acidosis, hyperammonemia, vomiting, lethargy, hepatomegaly | Developmental delay, motor deficits, potential for intellectual developmental disabilities |
| Trifunctional protein deficiency | Cardiovascular, pulmonary | Hypoglycemia, lactic acidosis, elevated creatine kinase, elevated liver transaminases, failure to thrive | Developmental delay, hypotonia, areflexia |
| Very long-chain acyl-CoA dehydrogenase deficiency | CNS, cardiovascular | Hypoketotic hypoglycemia, arrhythmias, lethargy, failure to thrive, cardiomyopathy, hepatomegaly | Developmental delay, motor deficits, potential for intellectual developmental disabilities |
CNS = central nervous system.
Adapted with permission from Waisbren SE. Expanded newborn screening: information and resources for the family physician. Am Fam Physician. 2008;77(7):991, with additional information from references 3, and 12 through 14.
Organic acid disorders (Table 33,11–13 ) represent particularly volatile conditions in which episodes of acidosis occur in the newborn period or later. Central nervous system abnormalities and developmental delay may occur despite treatment with special diets, supplements, or medications.
Table 3. Newborn Screening: Organic Acid Disorders
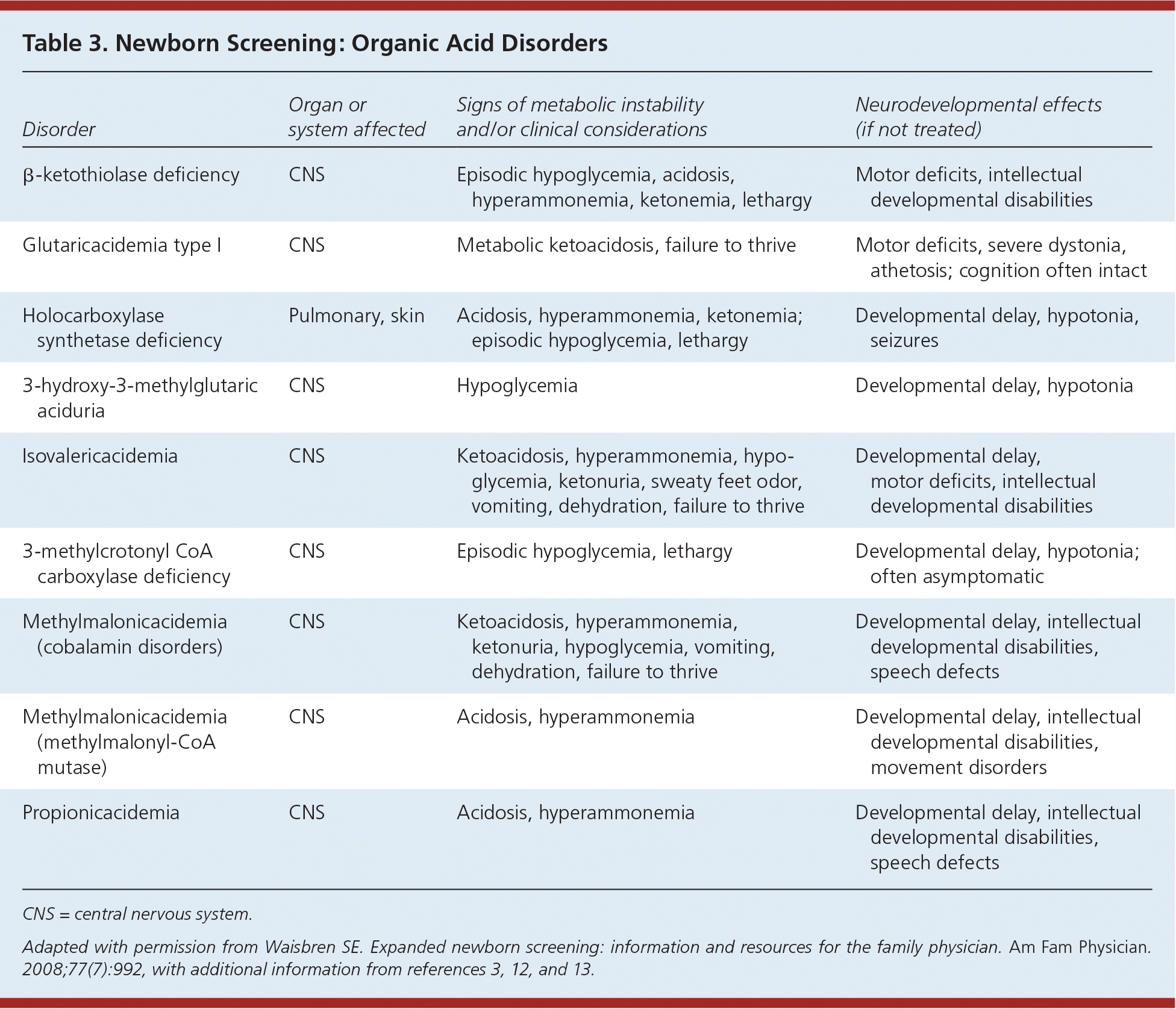
| Disorder | Organ or system affected | Signs of metabolic instability and/or clinical considerations | Neurodevelopmental effects (if not treated) |
|---|---|---|---|
| β-ketothiolase deficiency | CNS | Episodic hypoglycemia, acidosis, hyperammonemia, ketonemia, lethargy | Motor deficits, intellectual developmental disabilities |
| Glutaricacidemia type I | CNS | Metabolic ketoacidosis, failure to thrive | Motor deficits, severe dystonia, athetosis; cognition often intact |
| Holocarboxylase synthetase deficiency | Pulmonary, skin | Acidosis, hyperammonemia, ketonemia; episodic hypoglycemia, lethargy | Developmental delay, hypotonia, seizures |
| 3-hydroxy-3-methylglutaric aciduria | CNS | Hypoglycemia | Developmental delay, hypotonia |
| Isovalericacidemia | CNS | Ketoacidosis, hyperammonemia, hypoglycemia, ketonuria, sweaty feet odor, vomiting, dehydration, failure to thrive | Developmental delay, motor deficits, intellectual developmental disabilities |
| 3-methylcrotonyl CoA carboxylase deficiency | CNS | Episodic hypoglycemia, lethargy | Developmental delay, hypotonia; often asymptomatic |
| Methylmalonicacidemia (cobalamin disorders) | CNS | Ketoacidosis, hyperammonemia, ketonuria, hypoglycemia, vomiting, dehydration, failure to thrive | Developmental delay, intellectual developmental disabilities, speech defects |
| Methylmalonicacidemia (methylmalonyl-CoA mutase) | CNS | Acidosis, hyperammonemia | Developmental delay, intellectual developmental disabilities, movement disorders |
| Propionicacidemia | CNS | Acidosis, hyperammonemia | Developmental delay, intellectual developmental disabilities, speech defects |
CNS = central nervous system.
Adapted with permission from Waisbren SE. Expanded newborn screening: information and resources for the family physician. Am Fam Physician. 2008;77(7):992, with additional information from references 3, 12, and 13.
HEMOGLOBINOPATHIES
Hemoglobinopathies (Table 43,12,13 ) are not uncommon, with severity ranging from mild anemias to organ system damage. They may be the result of structural abnormalities in the hemoglobin molecule, such as sickle cell anemia, which is the most common inherited condition that is included in newborn screening. The clinical manifestations of hemoglobinopathies may also be caused by an inadequate production of hemoglobin due to α-thalassemia. Because of genetic intermingling, hemoglobinopathies can occur in any ethnic group, although they are more common in certain groups. Treatment includes support for persistent anemias, including blood transfusions; pain management; prophylactic antibiotics; vaccinations; and medical screenings to assess end-organ damage.
Table 4. Newborn Screening: Hemoglobinopathies

| Disorders | Organ or system affected | Signs of instability and/or clinical considerations | Neurodevelopmental effects (if not treated) |
|---|---|---|---|
| S, β-thalassemia, S,C disease, and S,S disease (sickle cell anemia) | Central nervous system, heme, kidney, lung | Anemia, infection, pain | Variable, mostly associated with increased risk of cerebrovascular accident |
ADDITIONAL DISORDERS
Tables 5 and 6 summarize endocrine disorders and miscellaneous multisystem diseases included in newborn screening.3,12,13 Cystic fibrosis is the second most common inherited life-shortening disease with childhood onset in the United States. Newborn screening evaluates functional defects in the cystic fibrosis protein. Biotinidase deficiency prevents the recycling of biotin, and patients with this disorder need daily biotin supplementation. The most common form of congenital adrenal hyperplasia is caused by 21-hydroxylase deficiency, which results in impaired adrenal synthesis of cortisol. Galactosemia is caused by deficiency of the galactose 1-phosphate uridyltransferase enzyme, which results in impaired galactose metabolism. The accumulation of galactose causes failure to thrive, infection, cataracts, liver failure, and death. Severe combined immunodeficiency is a primary immune deficiency syndrome with a severe defect in the T and B lymphocytes, causing increased susceptibility to infections. It is the first newborn screening disorder with identification based on DNA.
Table 5. Newborn Screening: Endocrine Disorders

| Disorder | Organ or system affected | Signs of instability and/or clinical considerations | Neurodevelopmental effects (if not treated) |
|---|---|---|---|
| Congenital adrenal hyperplasia | Adrenal, gastrointestinal, genitourinary, central nervous system | Poor feeding, vomiting, dehydration, hyponatremia | Intellectual developmental disabilities, learning disabilities, short stature, psychosexual disturbances |
| Primary congenital hypothyroidism | Thyroid, muscle, gastrointestinal | Jaundice, large fontanelles, pallor, myxedema | Delayed growth and weight gain; hypotonia; developmental delay |
Table 6. Newborn Screening: Miscellaneous Multisystem Diseases
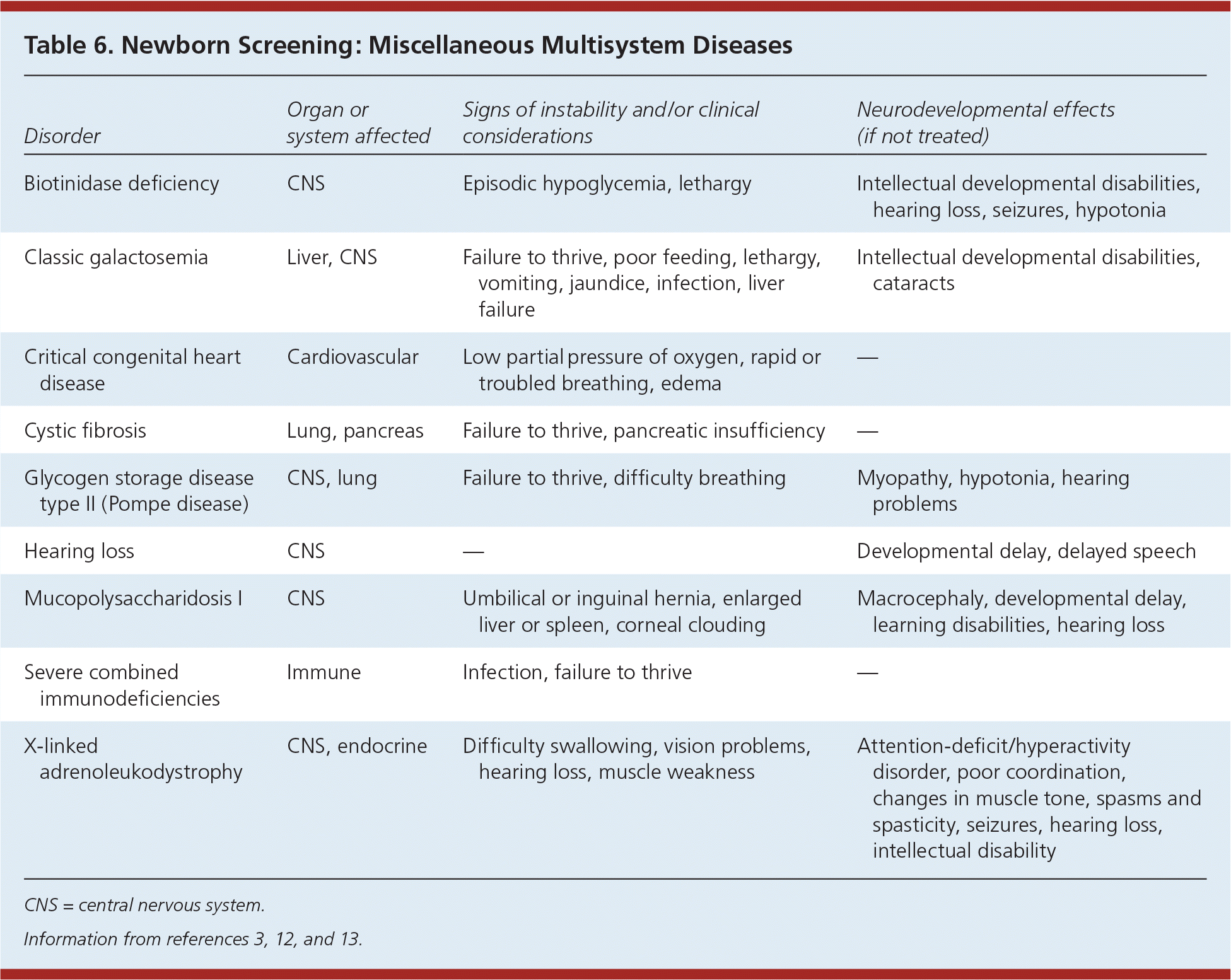
| Disorder | Organ or system affected | Signs of instability and/or clinical considerations | Neurodevelopmental effects (if not treated) |
|---|---|---|---|
| Biotinidase deficiency | CNS | Episodic hypoglycemia, lethargy | Intellectual developmental disabilities, hearing loss, seizures, hypotonia |
| Classic galactosemia | Liver, CNS | Failure to thrive, poor feeding, lethargy, vomiting, jaundice, infection, liver failure | Intellectual developmental disabilities, cataracts |
| Critical congenital heart disease | Cardiovascular | Low partial pressure of oxygen, rapid or troubled breathing, edema | — |
| Cystic fibrosis | Lung, pancreas | Failure to thrive, pancreatic insufficiency | — |
| Glycogen storage disease type II (Pompe disease) | CNS, lung | Failure to thrive, difficulty breathing | Myopathy, hypotonia, hearing problems |
| Hearing loss | CNS | — | Developmental delay, delayed speech |
| Mucopolysaccharidosis I | CNS | Umbilical or inguinal hernia, enlarged liver or spleen, corneal clouding | Macrocephaly, developmental delay, learning disabilities, hearing loss |
| Severe combined immunodeficiencies | Immune | Infection, failure to thrive | — |
| X-linked adrenoleukodystrophy | CNS, endocrine | Difficulty swallowing, vision problems, hearing loss, muscle weakness | Attention-deficit/hyperactivity disorder, poor coordination, changes in muscle tone, spasms and spasticity, seizures, hearing loss, intellectual disability |
CNS = central nervous system.
Mucopolysaccharidosis I and X-linked adrenoleukodystrophy were recently added to the Recommended Uniform Screening Panel. Mucopolysaccharidosis I is a lysosomal storage disease resulting in buildup of glycosaminoglycans in cells, blood, and connective tissues. The result is permanent, progressive cellular damage that affects appearance, physical abilities, organ and system functioning, and often mental development. X-linked adrenoleukodystrophy is a disorder of peroxisomal fatty acid beta oxidation that results in the accumulation of very long-chain fatty acids in tissues throughout the body. One of the most severely affected tissues is myelin in the central nervous system. Damage to the myelin sheaths of the nerves results in seizures and hyperactivity. Other adverse affects include problems with speaking, listening, and understanding verbal instructions.
Resources
The American College of Medical Genetics and Genomics has published resources to guide physicians if a newborn screening result is abnormal. For each marker(s), two items are provided: an action sheet that describes the short-term actions a clinician should follow when communicating with the family and determining the appropriate steps in follow-up, and an algorithm of the basic steps involved in determining the final diagnosis. These resources can be accessed at http://www.ncbi.nlm.nih.gov/books/NBK55827/.16
Detailed fact sheets on many disorders have been published in Pediatrics17 and are available free of charge at http://pediatrics.aappublications.org/content/118/3/e934.full.pdf+html?sid=8e387814-ea2b-4a20-817f-a923ca0eddde. Acute illness protocols are available from the New England Consortium of Metabolic Programs at http://newenglandconsortium.org/for-professionals/acute-illness-protocols/.
Follow-Up
Primary care physicians provide routine care for children with abnormal newborn screening results between visits for specialized medical care and treatment. Routine follow-up includes a standard neurologic and medical examination with specific attention to hepatomegaly, cardiovascular health, and neurologic function. A medical history is obtained, including information on hospitalizations, emergency department visits, medications, treatment, and developmental milestones. Results from blood tests such as liver function tests, electrolyte measurements, and complete blood count; cardiac studies, including echocardiography and electrocardiography; and an ophthalmologic examination can provide additional data on the associated effects of a disorder.
When an infant is ill or showing possible symptoms of metabolic crisis (e.g., lethargy, vomiting, ataxia), early contact with a metabolic subspecialist is important.18 Basic steps should be taken to stabilize the infant, including avoidance of the nutrient that cannot be metabolized, admittance to the emergency department, and prompt initiation of anticatabolic therapy.18
Developmental and neuropsychological evaluations are important for routine follow-up of children with metabolic disorders. When possible, psychologists familiar with metabolic disorders should perform the evaluation because they may be able to more easily recognize the subtle warning signs and symptoms associated with the disorder.11
Data Sources: A PubMed search was completed using the terms expanded newborn screening, neonatal screening methods, and metabolism inborn errors. The search included randomized controlled trials, meta-analyses, clinical trials, and clinical reviews. We also searched Essential Evidence Plus, the Cochrane Database of Systematic Reviews, the Agency for Healthcare Research and Quality Evidence Reports, and the U.S. Preventive Services Task Force. Search dates: November 1, 2015, and December 31, 2016.
The author thanks Carenado Davis, PhD, MLS, for assistance with the literature search.
